

Pan-cancer image-based detection of clinically actionable genetic alterations
- PMID: 33763651
- PMCID: PMC7610412
- DOI: 10.1038/s43018-020-0087-6
Pan-cancer image-based detection of clinically actionable genetic alterations
Erratum in
-
Author Correction: Pan-cancer image-based detection of clinically actionable genetic alterations.Nat Cancer. 2020 Nov;1(11):1129. doi: 10.1038/s43018-020-00149-6. Nat Cancer. 2020. PMID: 35122072 No abstract available.
Abstract
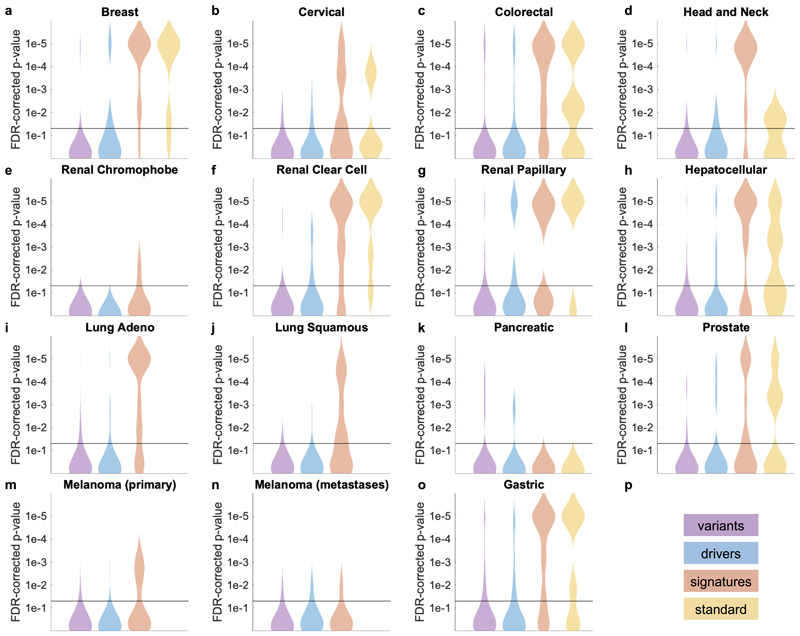
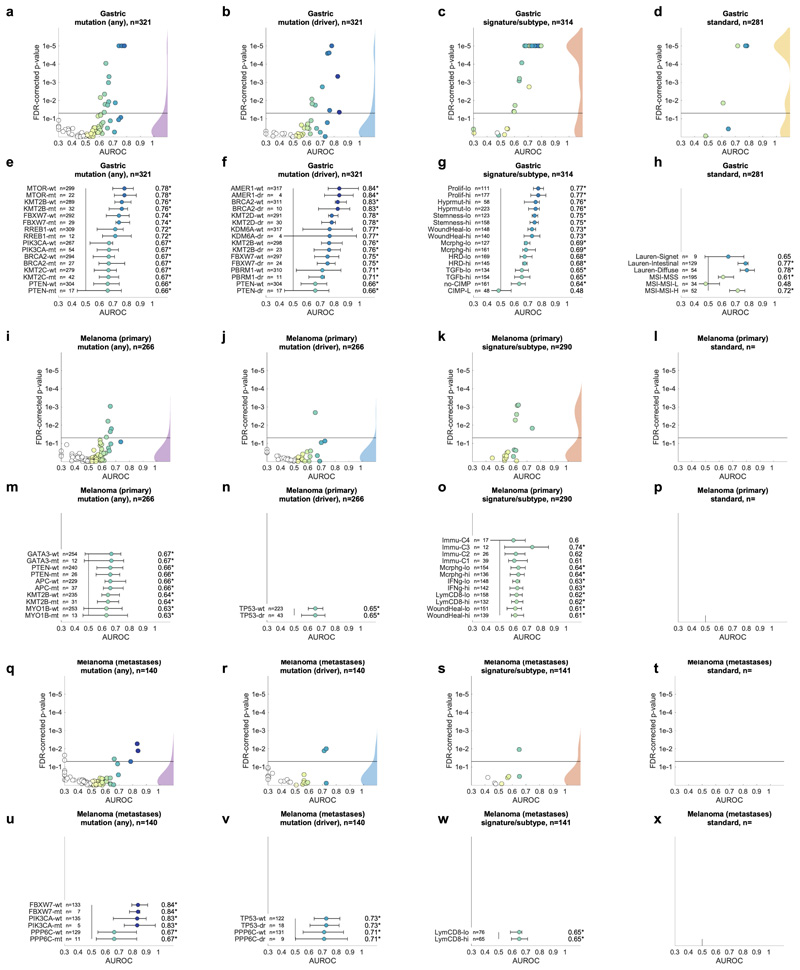
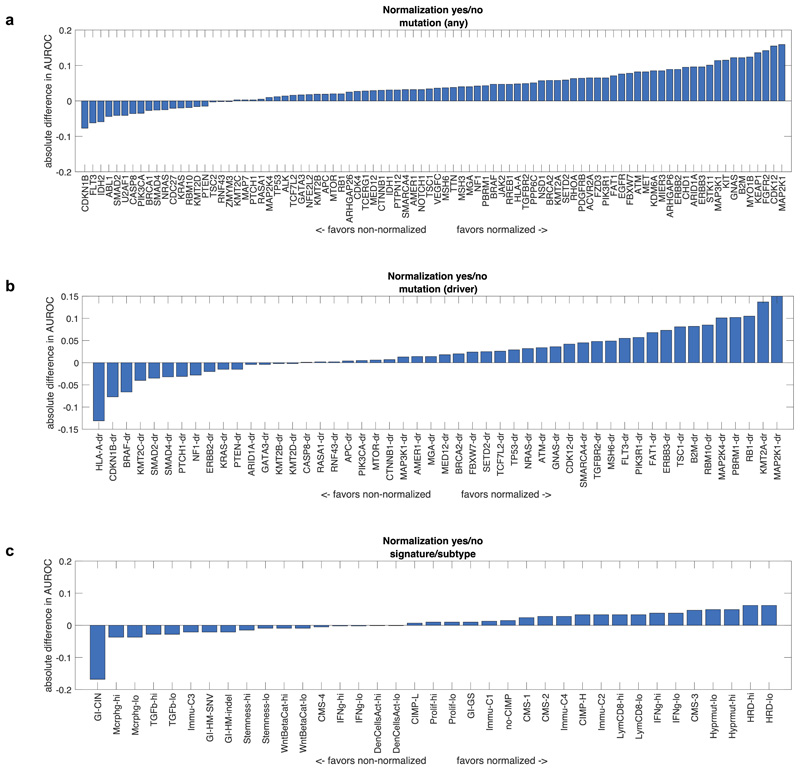
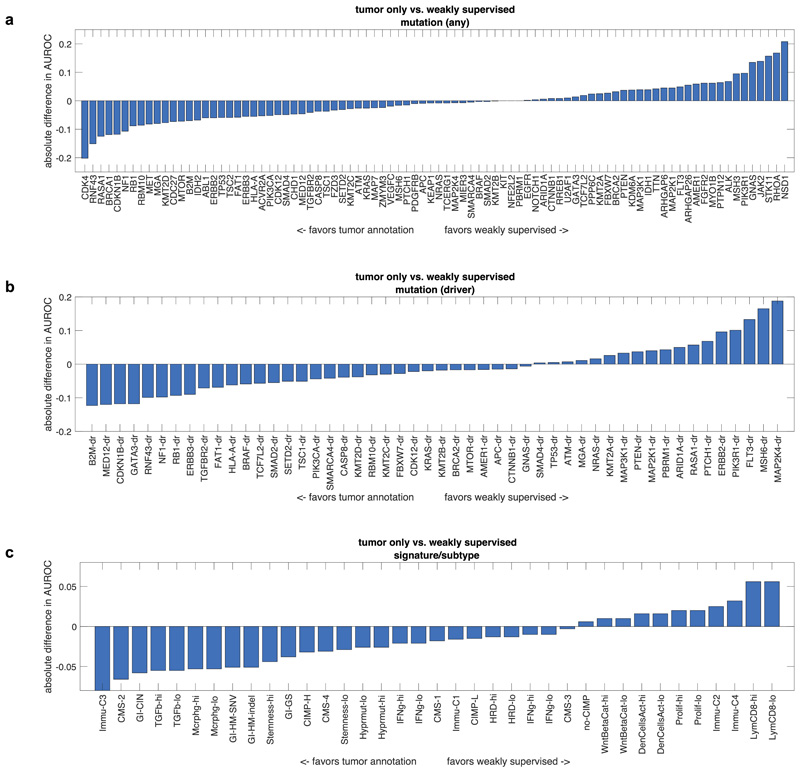
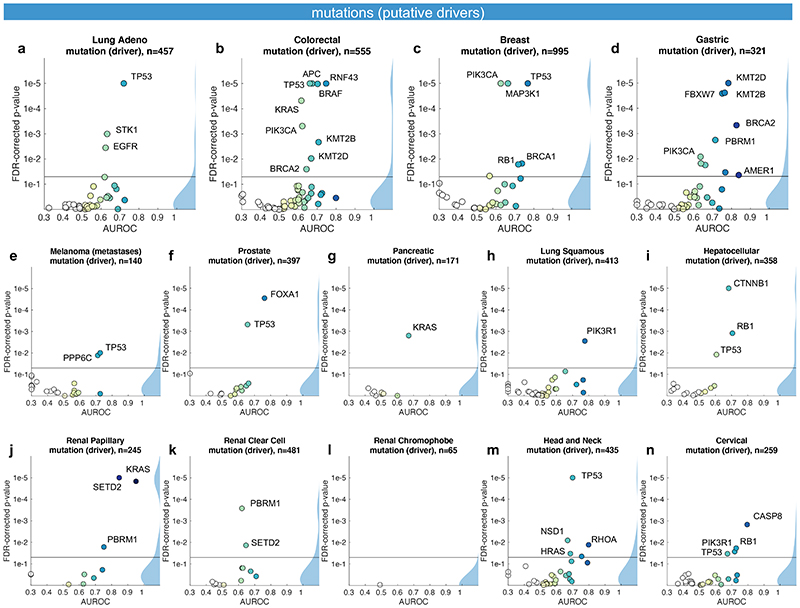
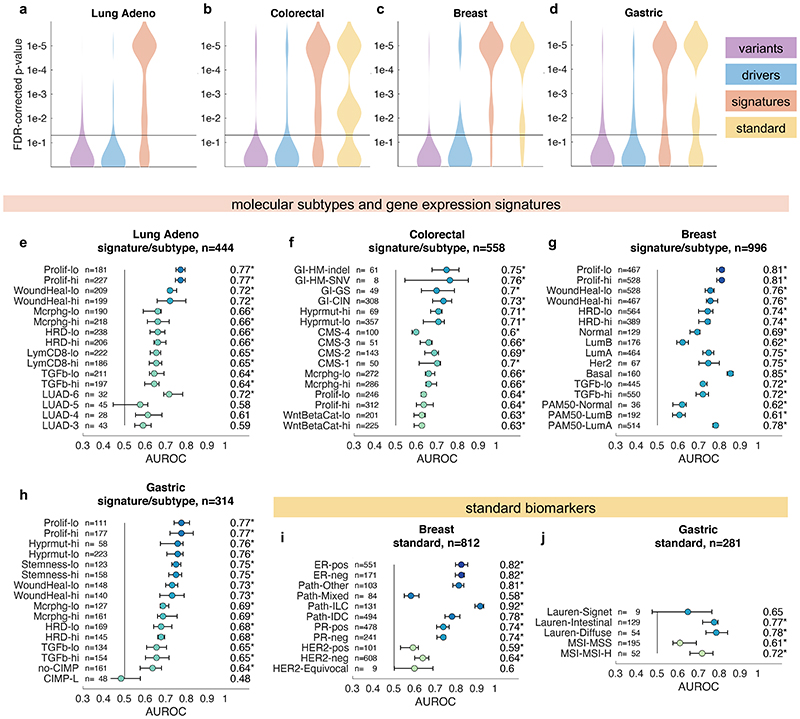
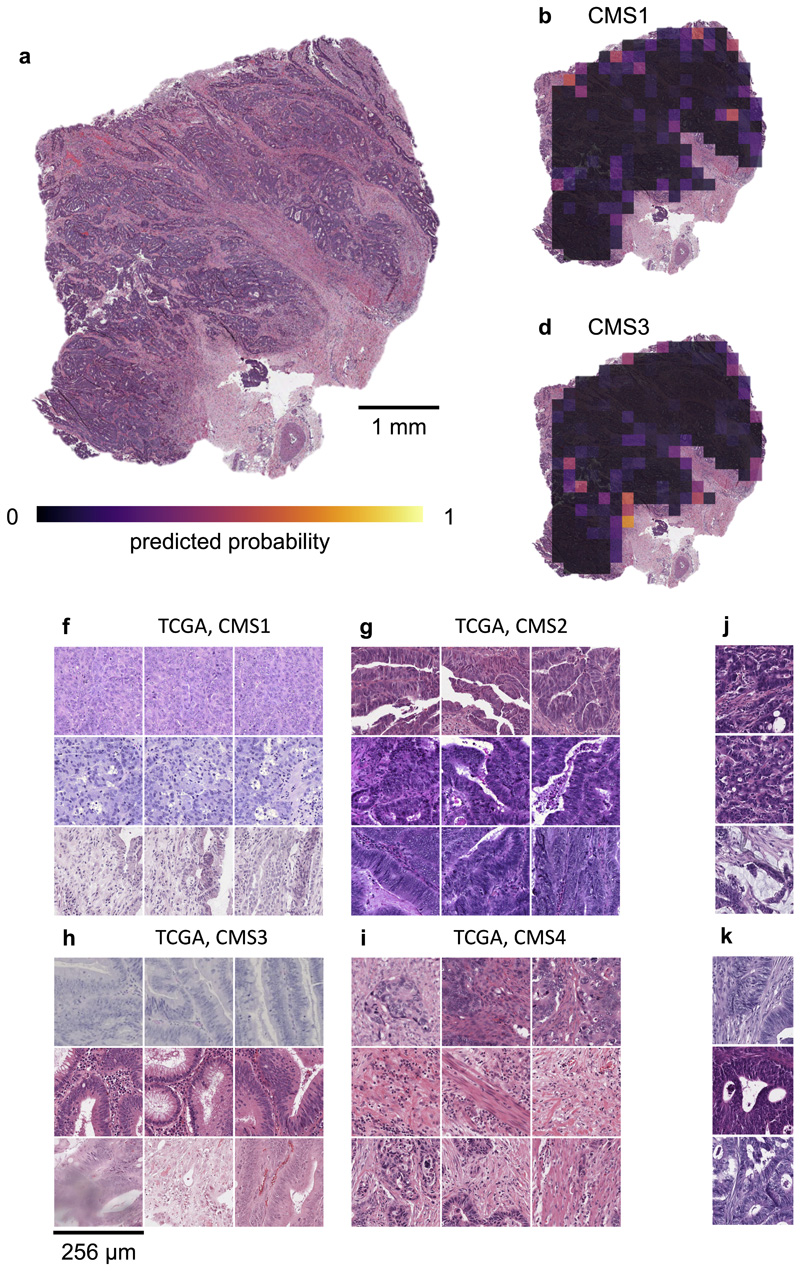
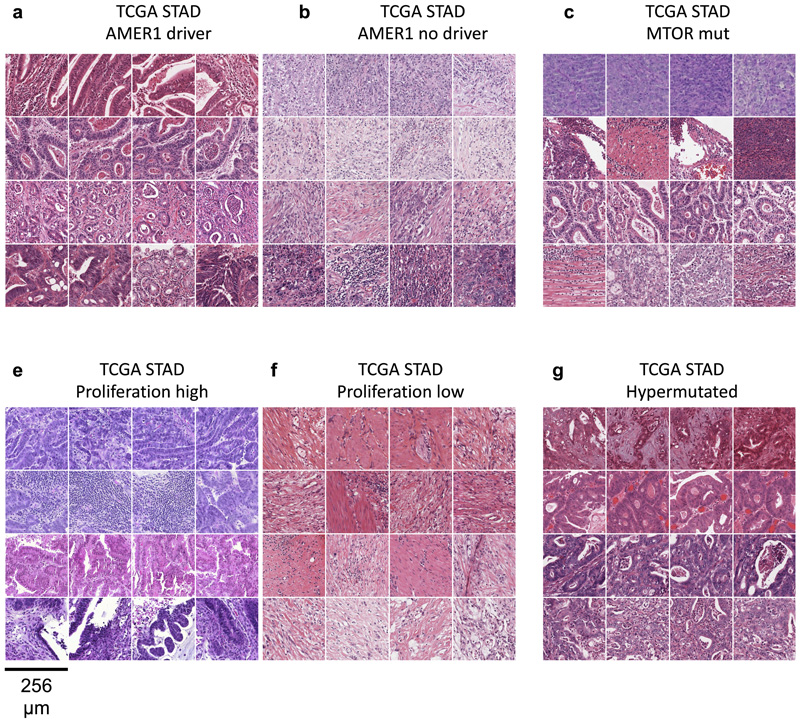
Molecular alterations in cancer can cause phenotypic changes in tumor cells and their micro-environment. Routine histopathology tissue slides - which are ubiquitously available - can reflect such morphological changes. Here, we show that deep learning can consistently infer a wide range of genetic mutations, molecular tumor subtypes, gene expression signatures and standard pathology biomarkers directly from routine histology. We developed, optimized, validated and publicly released a one-stop-shop workflow and applied it to tissue slides of more than 5000 patients across multiple solid tumors. Our findings show that a single deep learning algorithm can be trained to predict a wide range of molecular alterations from routine, paraffin-embedded histology slides stained with hematoxylin and eosin. These predictions generalize to other populations and are spatially resolved. Our method can be implemented on mobile hardware, potentially enabling point-of-care diagnostics for personalized cancer treatment. More generally, this approach could elucidate and quantify genotype-phenotype links in cancer.
Conflict of interest statement
Competing interests JNK has an informal, unpaid advisory role at Pathomix (Heidelberg, Germany) which does not relate to this research. JNK declares no other relationships or competing interests. All other authors declare no competing interests.
Figures




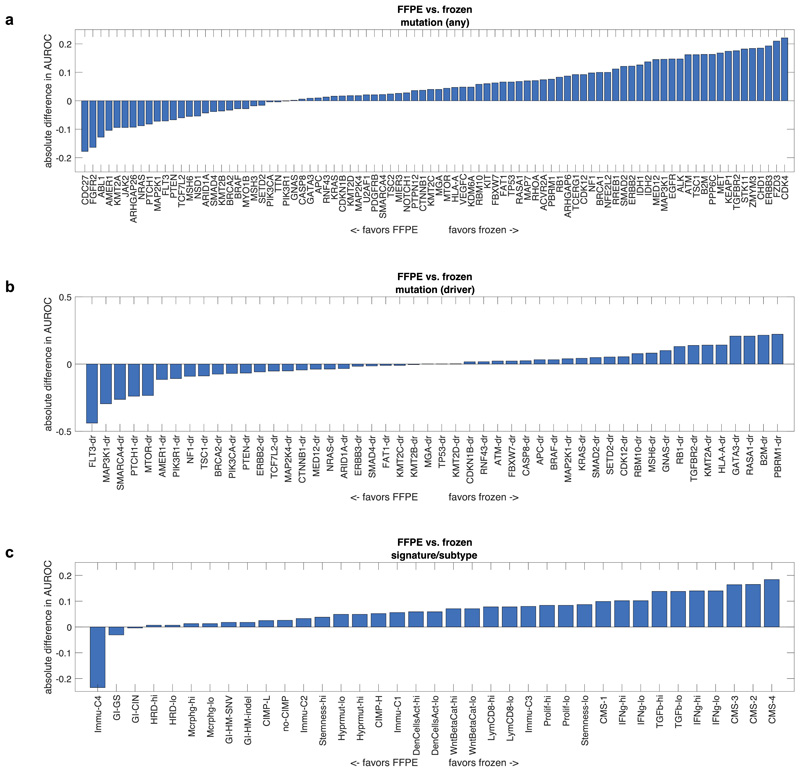
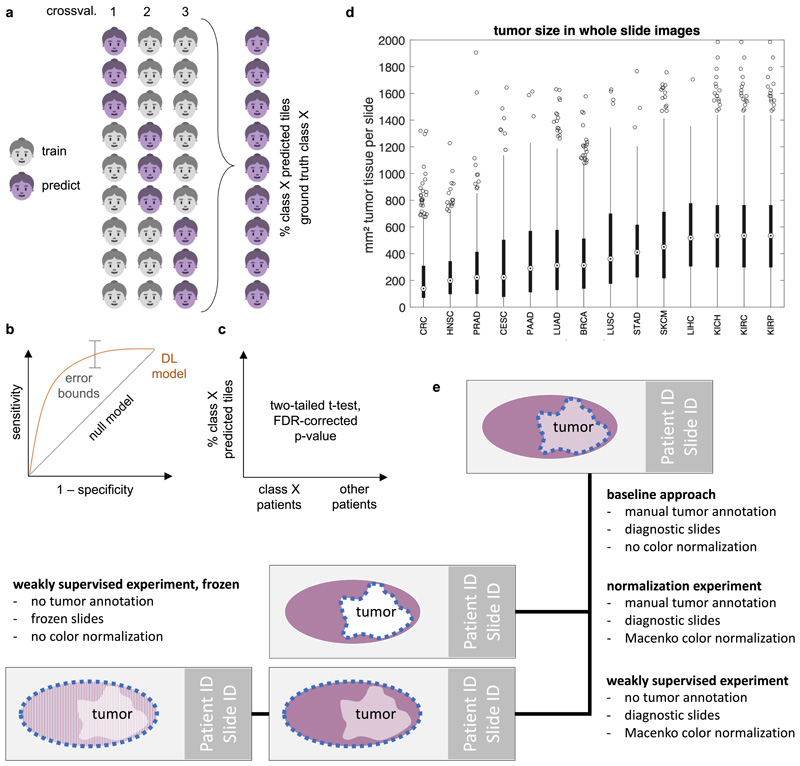
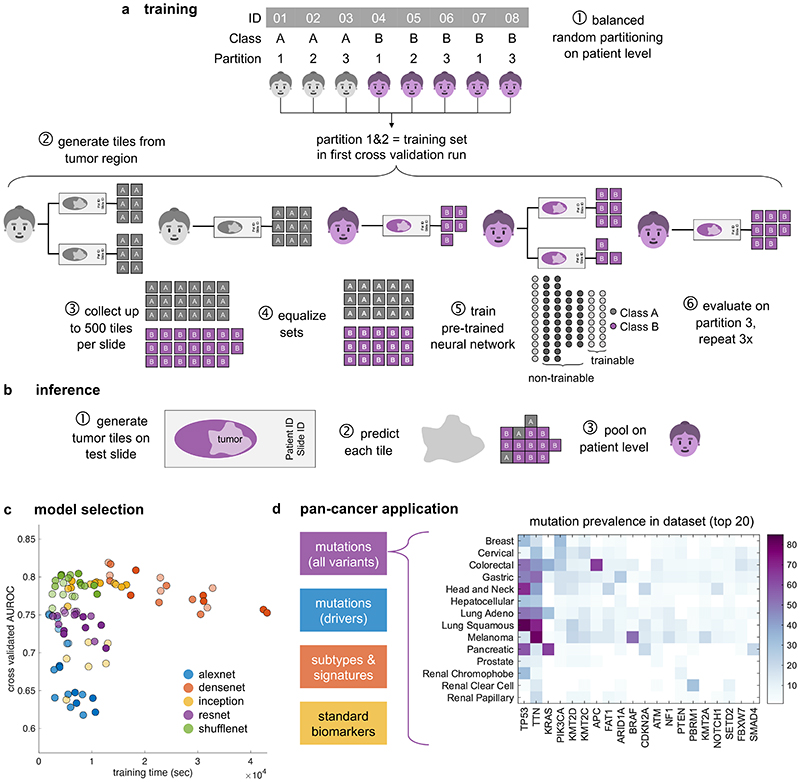
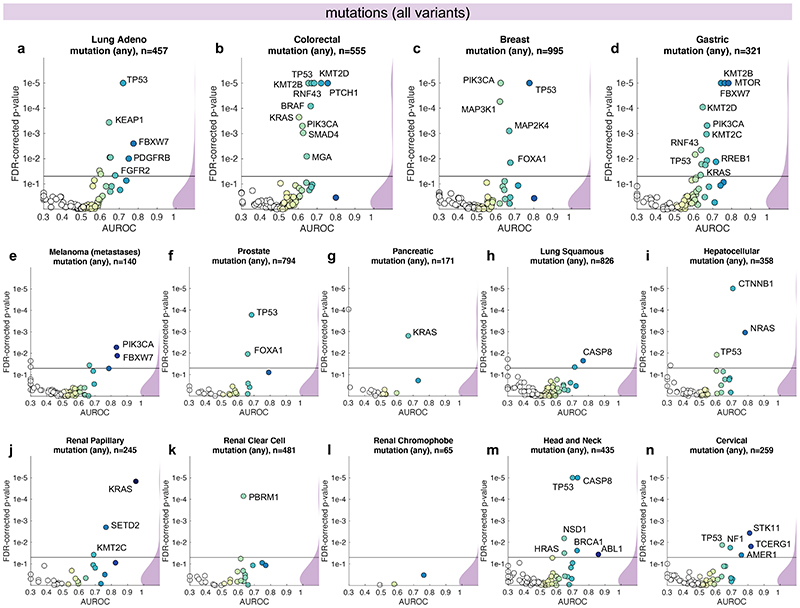
Comment in
-
Deep learning links histology, molecular signatures and prognosis in cancer.Nat Cancer. 2020 Aug;1(8):755-757. doi: 10.1038/s43018-020-0099-2. Nat Cancer. 2020. PMID: 35122048 Free PMC article.
References
-
- Kather JN, Halama N, Jaeger D. Genomics and emerging biomarkers for immunotherapy of colorectal cancer. Seminars in Cancer Biology. 2018;52:189–197. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
