

Toward a broader view of mechanisms of drug cardiotoxicity
- PMID: 33763655
- PMCID: PMC7974548
- DOI: 10.1016/j.xcrm.2021.100216
Toward a broader view of mechanisms of drug cardiotoxicity
Abstract
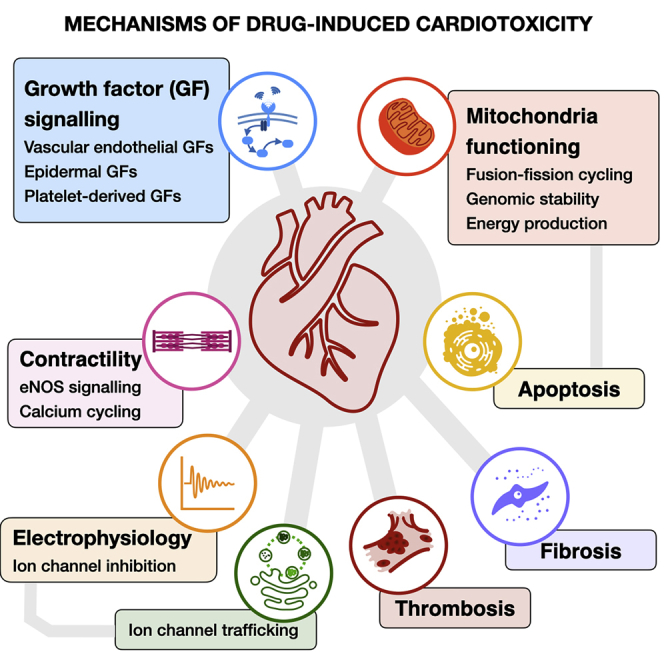
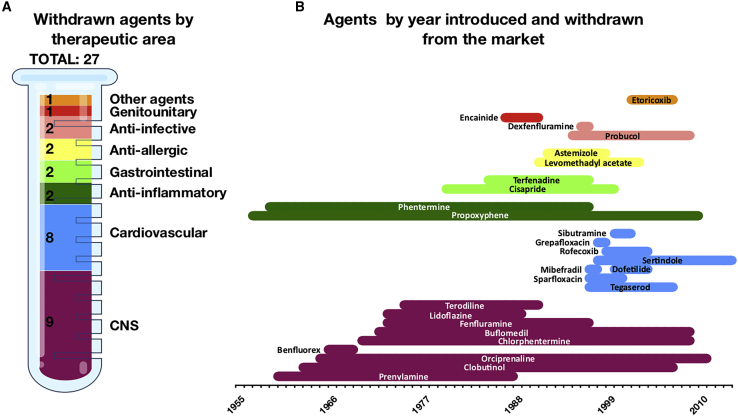
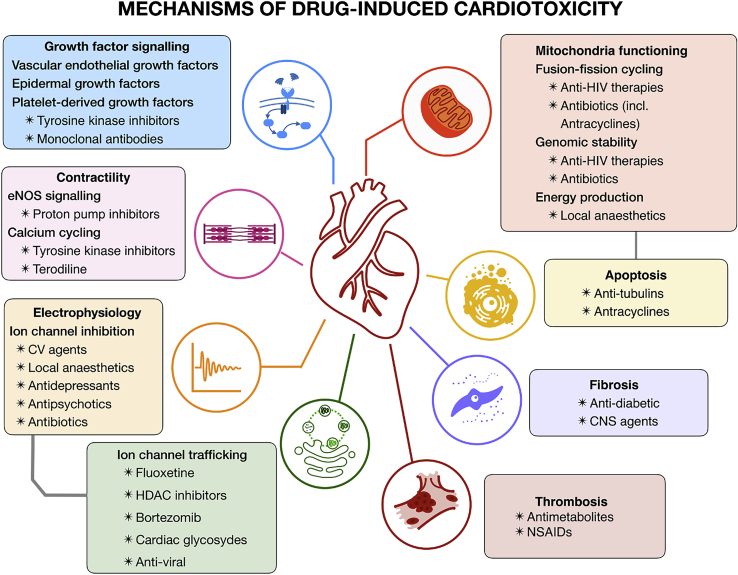
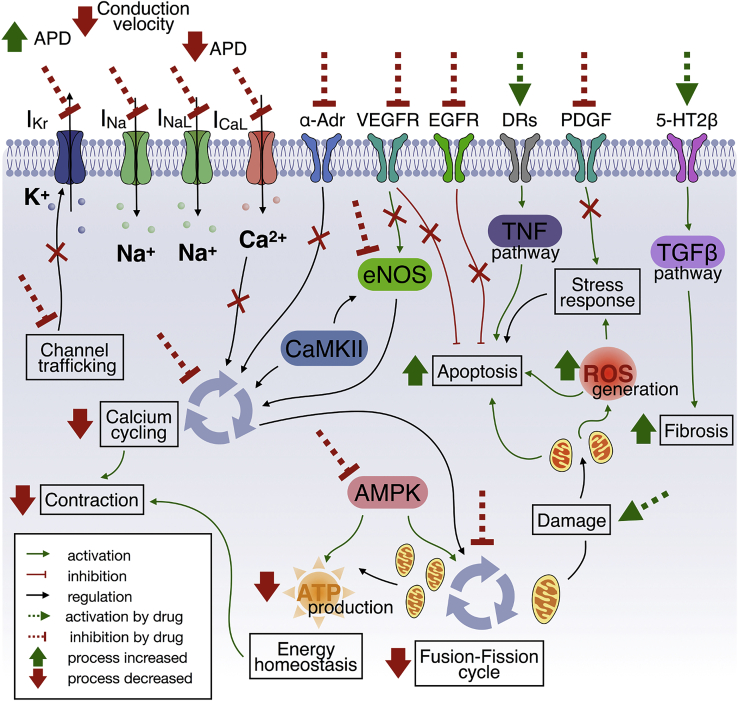
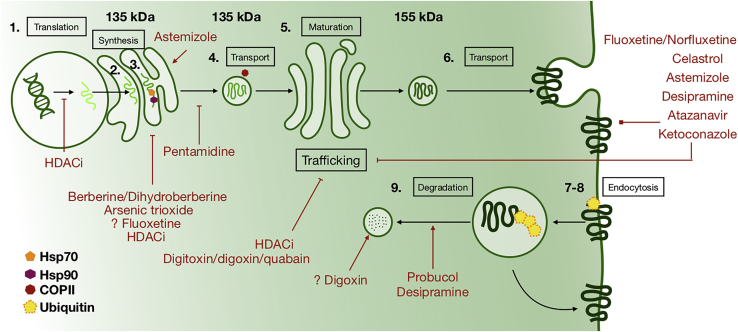
Cardiotoxicity, defined as toxicity that affects the heart, is one of the most common adverse drug effects. Numerous drugs have been shown to have the potential to induce lethal arrhythmias by affecting cardiac electrophysiology, which is the focus of current preclinical testing. However, a substantial number of drugs can also affect cardiac function beyond electrophysiology. Within this broader sense of cardiotoxicity, this review discusses the key drug-protein interactions known to be involved in cardiotoxic drug response. We cover adverse effects of anticancer, central nervous system, genitourinary system, gastrointestinal, antihistaminic, anti-inflammatory, and anti-infective agents, illustrating that many share mechanisms of cardiotoxicity, including contractility, mitochondrial function, and cellular signaling.
Keywords: adverse reactions; cardiotoxicity; cell signaling; mechanisms of toxicity; side effects.
© 2021 The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Ferri N., Siegl P., Corsini A., Herrmann J., Lerman A., Benghozi R. Drug attrition during pre-clinical and clinical development: understanding and managing drug-induced cardiotoxicity. Pharmacol. Ther. 2013;138:470–484. - PubMed
-
- Sloan J.A., Goldberg R.M., Sargent D.J., Vargas-Chanes D., Nair S., Cha S.S., Novotny P.J., Poon M.A., O’Connell M.J., Loprinzi C.L. Women experience greater toxicity with fluorouracil-based chemotherapy for colorectal cancer. J. Clin. Oncol. 2002;20:1491–1498. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
