

Improved SILAC Quantification with Data-Independent Acquisition to Investigate Bortezomib-Induced Protein Degradation
- PMID: 33764077
- PMCID: PMC8256668
- DOI: 10.1021/acs.jproteome.0c00938
Improved SILAC Quantification with Data-Independent Acquisition to Investigate Bortezomib-Induced Protein Degradation
Abstract
Stable isotope labeling by amino acids in cell culture (SILAC) coupled to data-dependent acquisition (DDA) is a common approach to quantitative proteomics with the desirable benefit of reducing batch effects during sample processing and data acquisition. More recently, using data-independent acquisition (DIA/SWATH) to systematically measure peptides has gained popularity for its comprehensiveness, reproducibility, and accuracy of quantification. The complementary advantages of these two techniques logically suggests combining them. Here we develop a SILAC-DIA-MS workflow using free, open-source software. We empirically determine that using DIA achieves similar peptide detection numbers as DDA and that DIA improves the quantitative accuracy and precision of SILAC by an order of magnitude. Finally, we apply SILAC-DIA-MS to determine protein turnover rates of cells treated with bortezomib, an FDA-approved 26S proteasome inhibitor for multiple myeloma and mantle cell lymphoma. We observe that SILAC-DIA produces more sensitive protein turnover models. Of the proteins determined to be differentially degraded by both acquisition methods, we find known proteins that are degraded by the ubiquitin-proteasome pathway, such as HNRNPK, EIF3A, and IF4A1/EIF4A-1, and a slower turnover for CATD, a protein implicated in invasive breast cancer. With improved quantification from DIA, we anticipate that this workflow will make SILAC-based experiments like protein turnover more sensitive.
Keywords: data independent acquisition; protein degradation; protein turnover; pulse SILAC; quantitative proteomics.
Figures
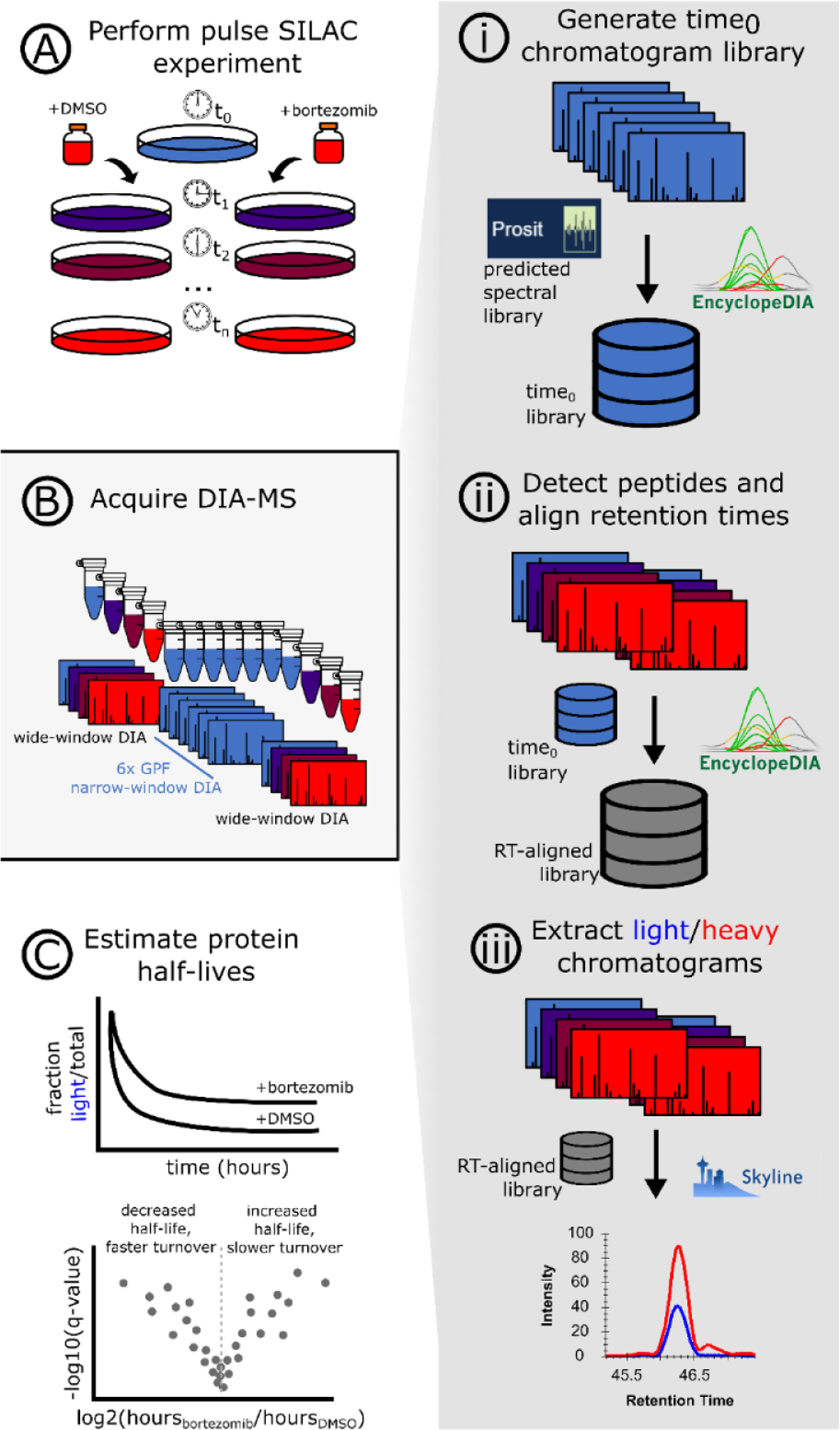
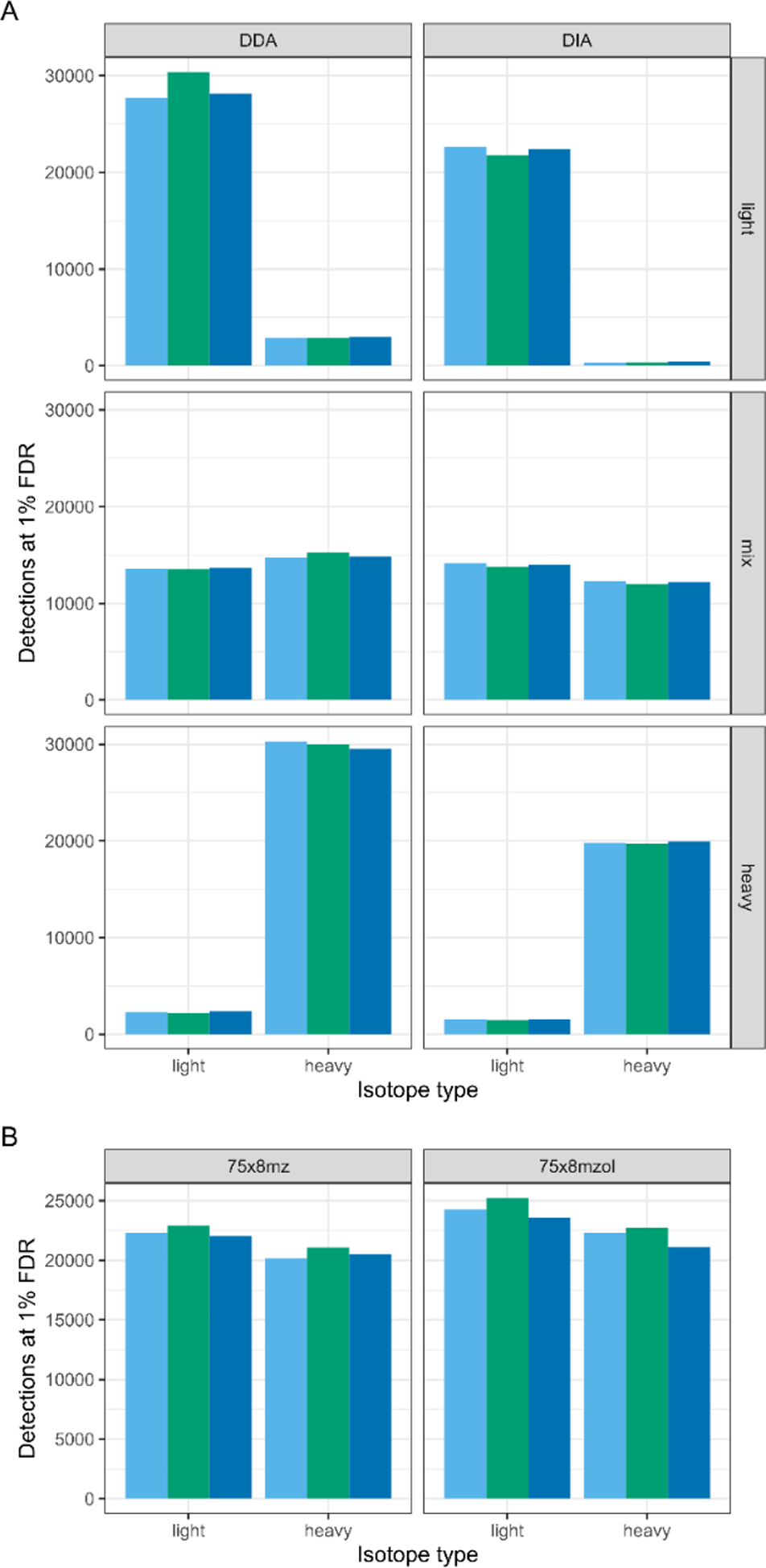
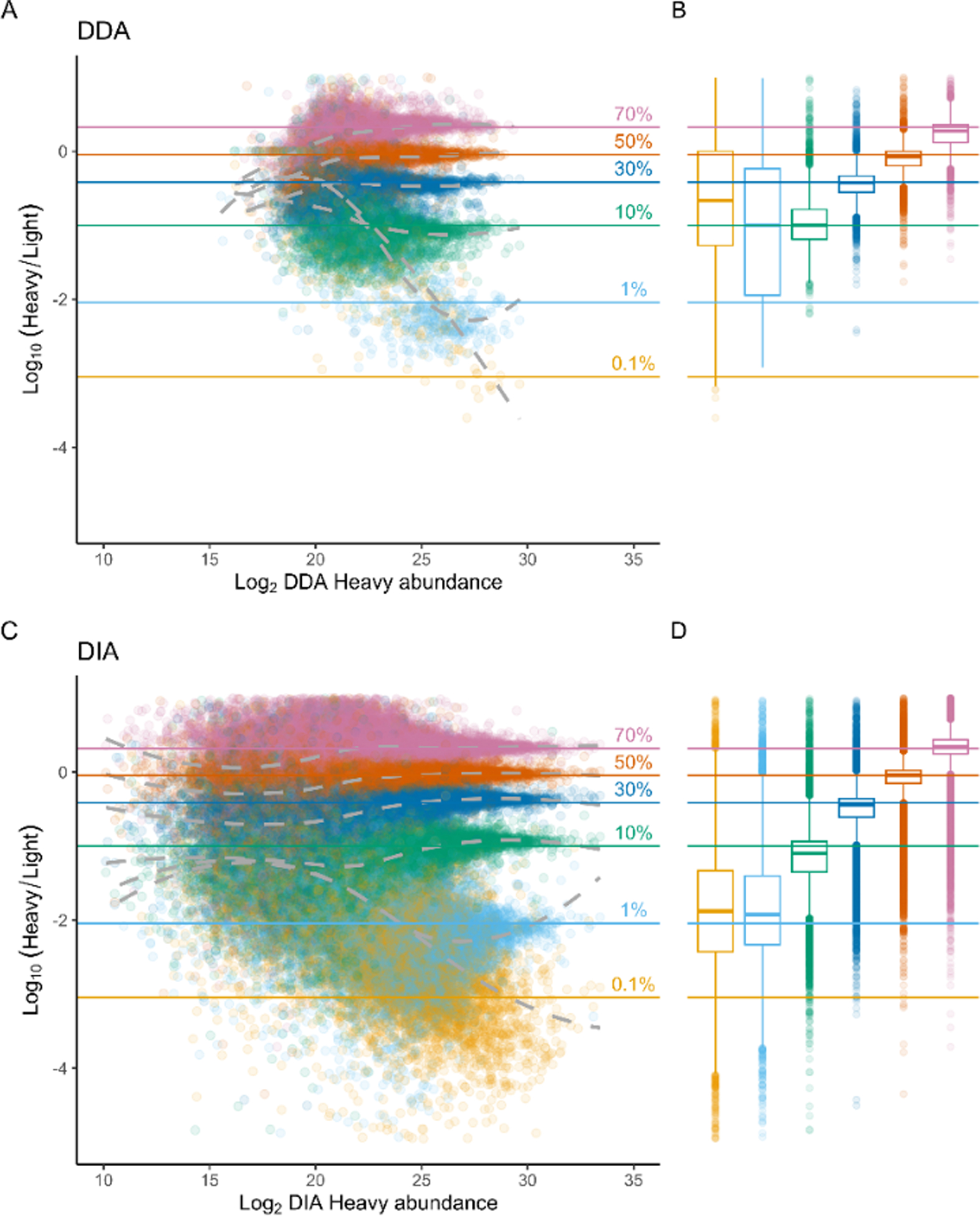
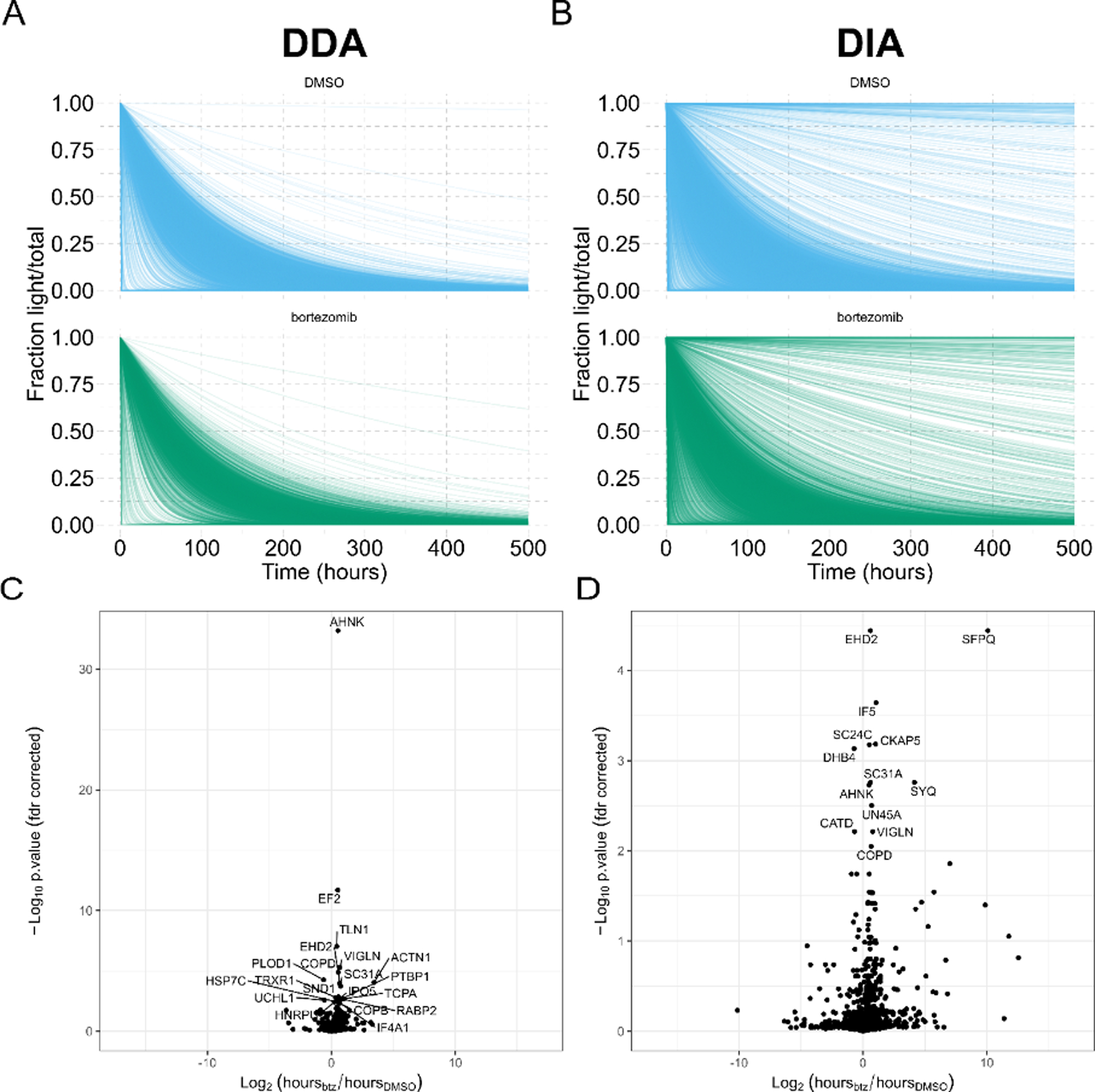
References
-
- Bantscheff M, Schirle M, Sweetman G, Rick J & Kuster B Quantitative mass spectrometry in proteomics: a critical review. Anal. Bioanal. Chem 389, 1017–1031 (2007). - PubMed
-
- Ong S-E, Kratchmarova I & Mann M Properties of 13 C-Substituted Arginine in Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC). J. Proteome Res 2, 173–181 (2003). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
