

Viral infection of cells within the tumor microenvironment mediates antitumor immunotherapy via selective TBK1-IRF3 signaling
- PMID: 33767151
- PMCID: PMC7994570
- DOI: 10.1038/s41467-021-22088-1
Viral infection of cells within the tumor microenvironment mediates antitumor immunotherapy via selective TBK1-IRF3 signaling
Abstract
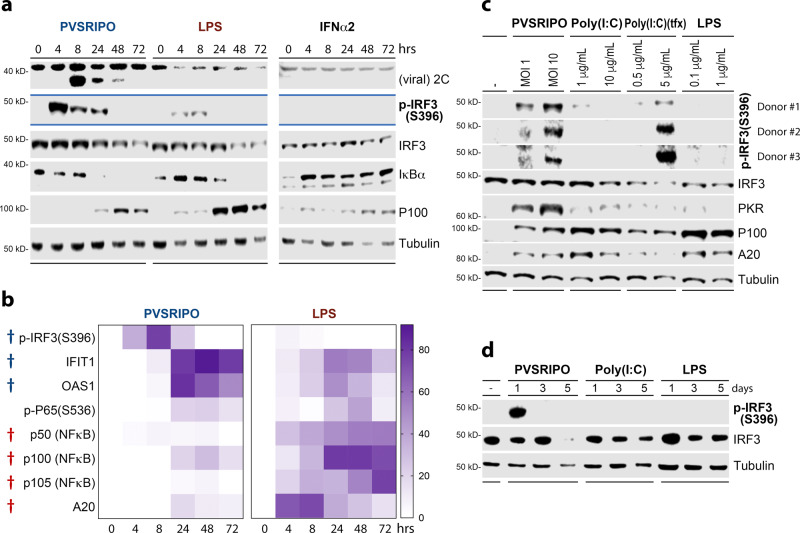
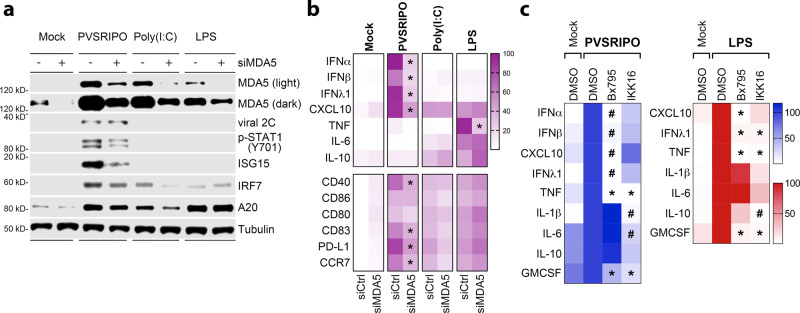
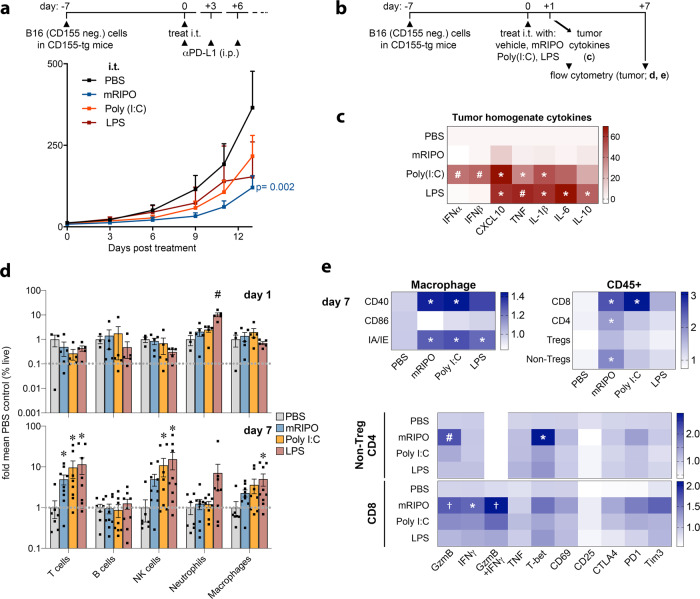
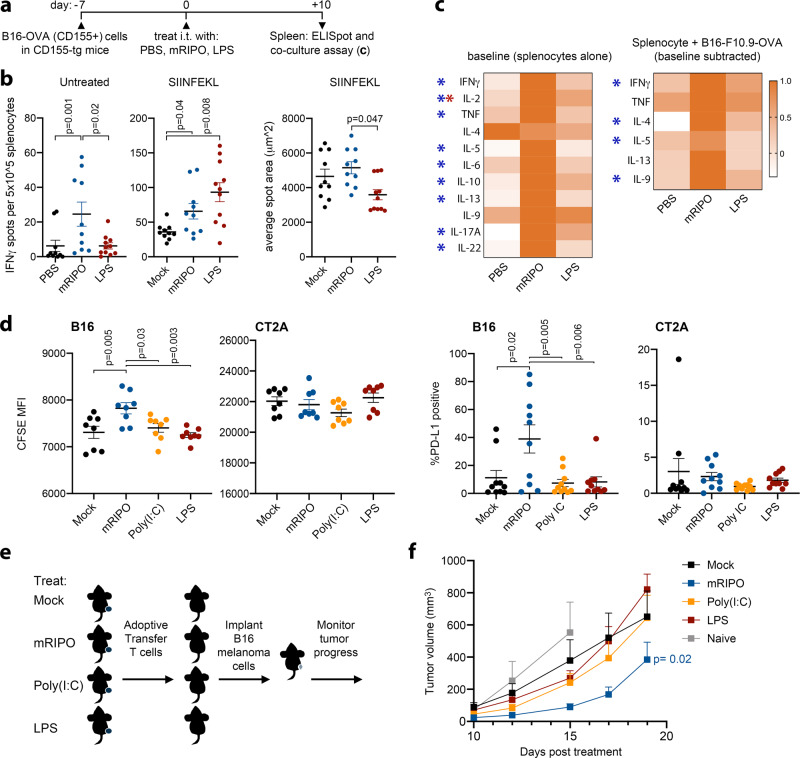
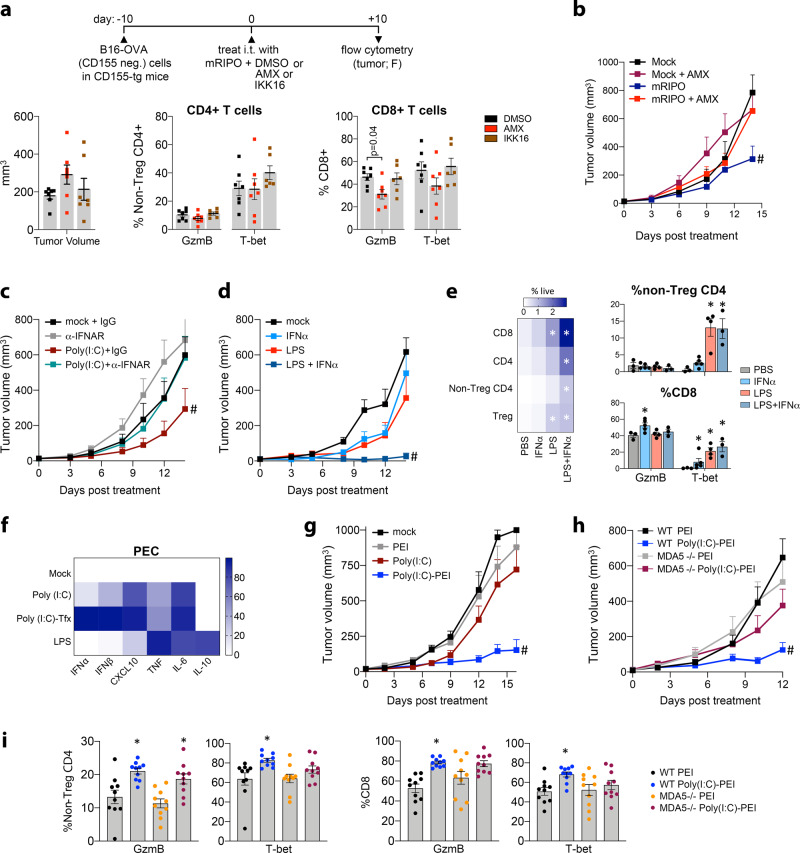
Activating intra-tumor innate immunity might enhance tumor immune surveillance. Virotherapy is proposed to achieve tumor cell killing, while indirectly activating innate immunity. Here, we report that recombinant poliovirus therapy primarily mediates antitumor immunotherapy via direct infection of non-malignant tumor microenvironment (TME) cells, independent of malignant cell lysis. Relative to other innate immune agonists, virotherapy provokes selective, TBK1-IRF3 driven innate inflammation that is associated with sustained type-I/III interferon (IFN) release. Despite priming equivalent antitumor T cell quantities, MDA5-orchestrated TBK1-IRF3 signaling, but not NFκB-polarized TLR activation, culminates in polyfunctional and Th1-differentiated antitumor T cell phenotypes. Recombinant type-I IFN increases tumor-localized T cell function, but does not mediate durable antitumor immunotherapy without concomitant pattern recognition receptor (PRR) signaling. Thus, virus-induced MDA5-TBK1-IRF3 signaling in the TME provides PRR-contextualized IFN responses that elicit functional antitumor T cell immunity. TBK1-IRF3 innate signal transduction stimulates eventual function and differentiation of tumor-infiltrating T cells.
Conflict of interest statement
M.C.B., D.D.B., D.M.A., S.K.N., and M.G. own intellectual property related to this research, which has been licensed to Istari Oncology, Inc. M.G. and D.D.B. are compensated advisors to- and own equity in Istari Oncology, Inc. S.K.N., M.C.B., D.D.B., and M.G. are inventors on patent application PCT/US2017/039953 held/submitted by Duke University that covers the composition and methods for activating antigen-presenting cells with PVSRIPO. All other authors declare no competing interests.
Figures
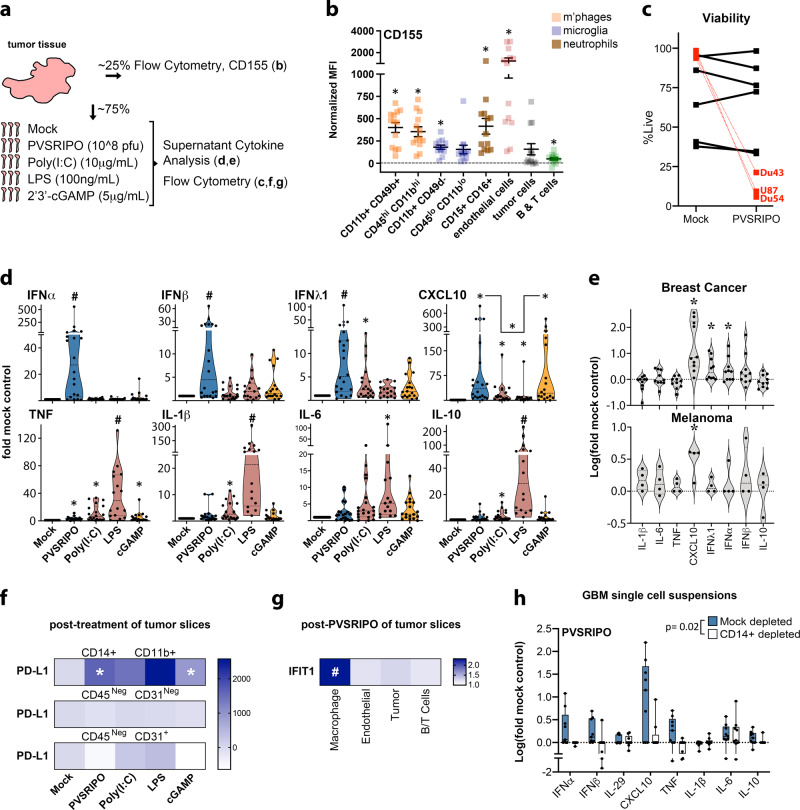
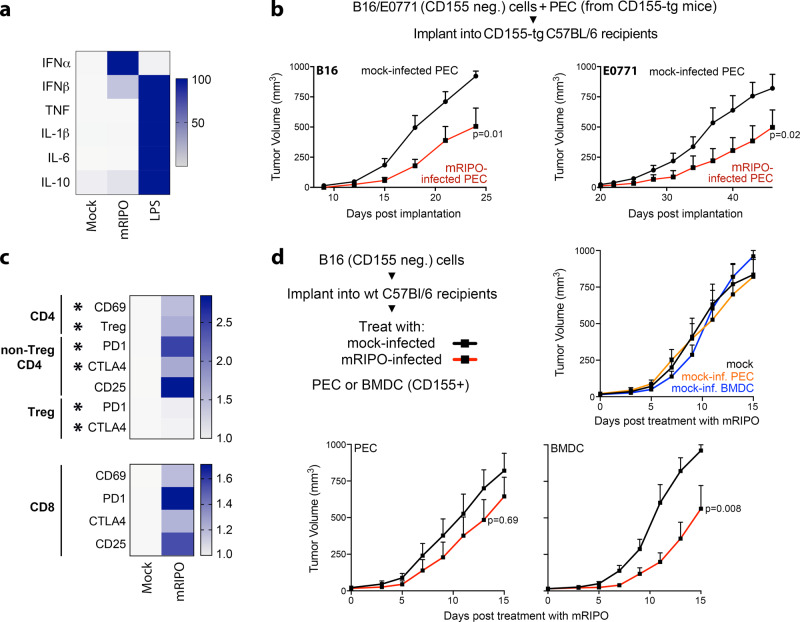
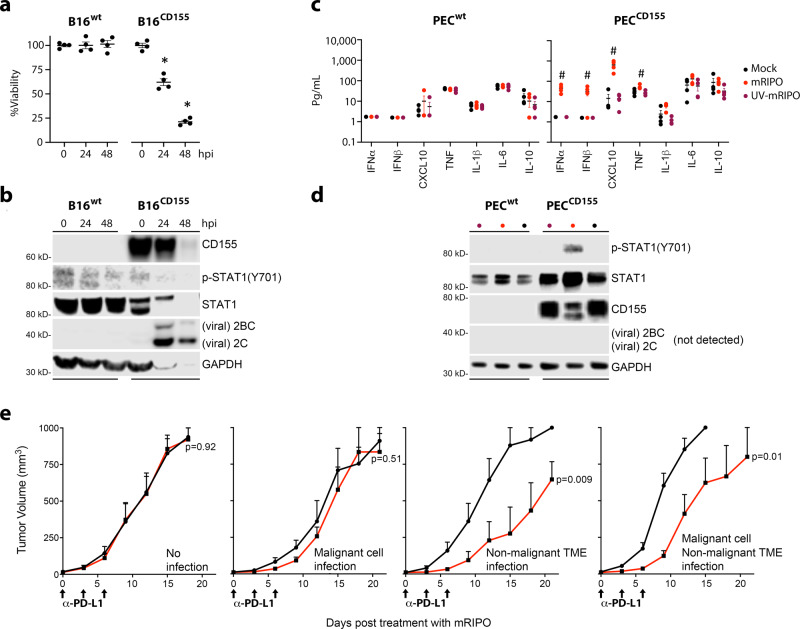
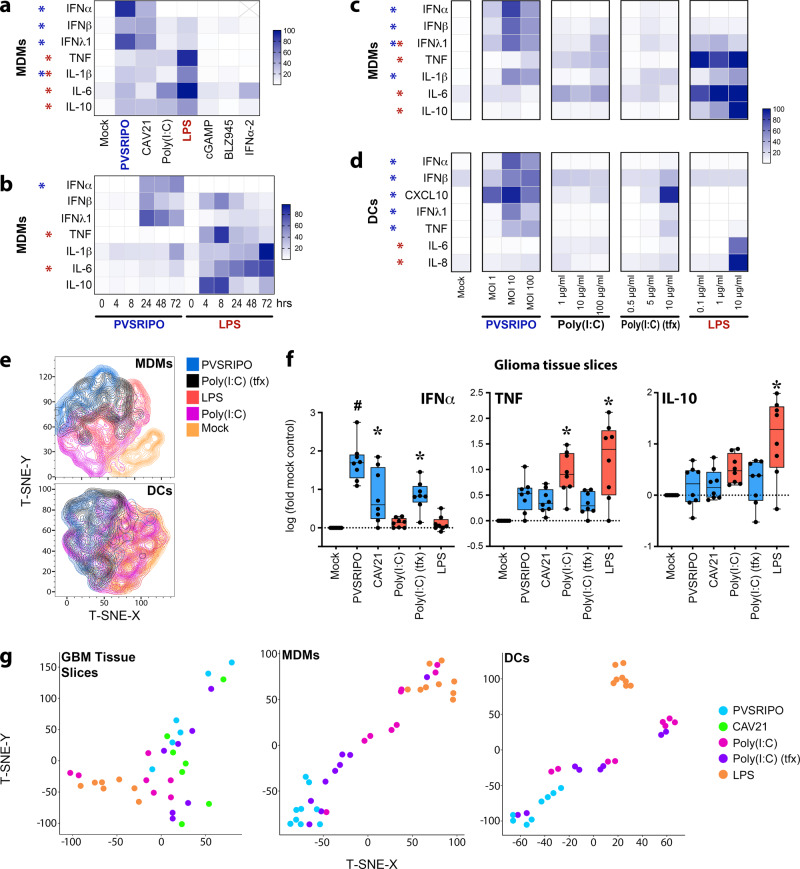
Similar articles
-
Heartland virus NSs protein disrupts host defenses by blocking the TBK1 kinase-IRF3 transcription factor interaction and signaling required for interferon induction.J Biol Chem. 2017 Oct 6;292(40):16722-16733. doi: 10.1074/jbc.M117.805127. Epub 2017 Aug 28. J Biol Chem. 2017. PMID: 28848048 Free PMC article.
-
Helicase-Driven Activation of NFκB-COX2 Pathway Mediates the Immunosuppressive Component of dsRNA-Driven Inflammation in the Human Tumor Microenvironment.Cancer Res. 2018 Aug 1;78(15):4292-4302. doi: 10.1158/0008-5472.CAN-17-3985. Epub 2018 May 31. Cancer Res. 2018. PMID: 29853604 Free PMC article.
-
The methyltransferase PRMT6 attenuates antiviral innate immunity by blocking TBK1-IRF3 signaling.Cell Mol Immunol. 2019 Oct;16(10):800-809. doi: 10.1038/s41423-018-0057-4. Epub 2018 Jul 4. Cell Mol Immunol. 2019. PMID: 29973649 Free PMC article.
-
Negative regulation of TBK1-mediated antiviral immunity.FEBS Lett. 2013 Mar 18;587(6):542-8. doi: 10.1016/j.febslet.2013.01.052. Epub 2013 Feb 8. FEBS Lett. 2013. PMID: 23395611 Free PMC article. Review.
-
The Role of Optineurin in Antiviral Type I Interferon Production.Front Immunol. 2018 Apr 26;9:853. doi: 10.3389/fimmu.2018.00853. eCollection 2018. Front Immunol. 2018. PMID: 29755463 Free PMC article. Review.
Cited by
-
Polio virotherapy targets the malignant glioma myeloid infiltrate with diffuse microglia activation engulfing the CNS.Neuro Oncol. 2023 Sep 5;25(9):1631-1643. doi: 10.1093/neuonc/noad052. Neuro Oncol. 2023. PMID: 36864784 Free PMC article.
-
Engaging Pattern Recognition Receptors in Solid Tumors to Generate Systemic Antitumor Immunity.Cancer Treat Res. 2022;183:91-129. doi: 10.1007/978-3-030-96376-7_3. Cancer Treat Res. 2022. PMID: 35551657
-
Type I interferon-mediated tumor immunity and its role in immunotherapy.Cell Mol Life Sci. 2022 Mar 16;79(3):191. doi: 10.1007/s00018-022-04219-z. Cell Mol Life Sci. 2022. PMID: 35292881 Free PMC article. Review.
-
The Role of Long Noncoding RNA BST2-2 in the Innate Immune Response to Viral Infection.J Virol. 2022 Apr 13;96(7):e0020722. doi: 10.1128/jvi.00207-22. Epub 2022 Mar 17. J Virol. 2022. PMID: 35297670 Free PMC article.
-
Focus on current and emerging treatment options for glioma: A comprehensive review.World J Clin Oncol. 2024 Apr 24;15(4):482-495. doi: 10.5306/wjco.v15.i4.482. World J Clin Oncol. 2024. PMID: 38689623 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
