

Deep viral blood metagenomics reveals extensive anellovirus diversity in healthy humans
- PMID: 33767340
- PMCID: PMC7994813
- DOI: 10.1038/s41598-021-86427-4
Deep viral blood metagenomics reveals extensive anellovirus diversity in healthy humans
Abstract
Human blood metagenomics has revealed the presence of different types of viruses in apparently healthy subjects. By far, anelloviruses constitute the viral family that is more frequently found in human blood, although amplification biases and contaminations pose a major challenge in this field. To investigate this further, we subjected pooled plasma samples from 120 healthy donors in Spain to high-speed centrifugation, RNA and DNA extraction, random amplification, and massive parallel sequencing. Our results confirm the extensive presence of anelloviruses in such samples, which represented nearly 97% of the total viral sequence reads obtained. We assembled 114 different viral genomes belonging to this family, revealing remarkable diversity. Phylogenetic analysis of ORF1 suggested 28 potentially novel anellovirus species, 24 of which were validated by Sanger sequencing to discard artifacts. These findings underscore the importance of implementing more efficient purification procedures that enrich the viral fraction as an essential step in virome studies and question the suggested pathological role of anelloviruses.
Conflict of interest statement
The authors declare no competing interests.
Figures
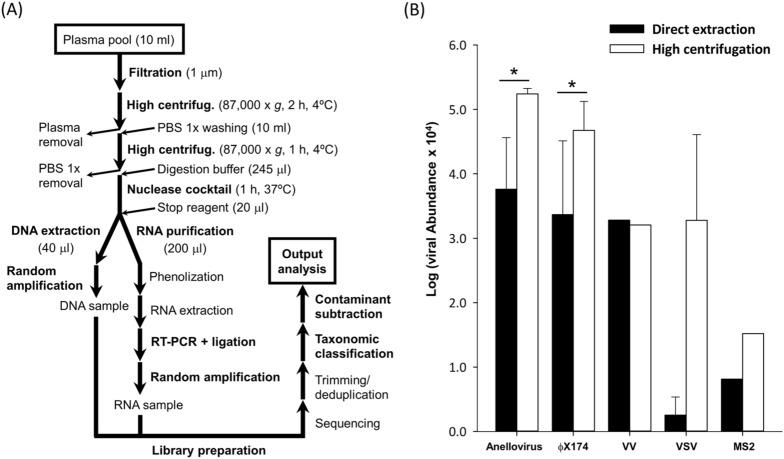
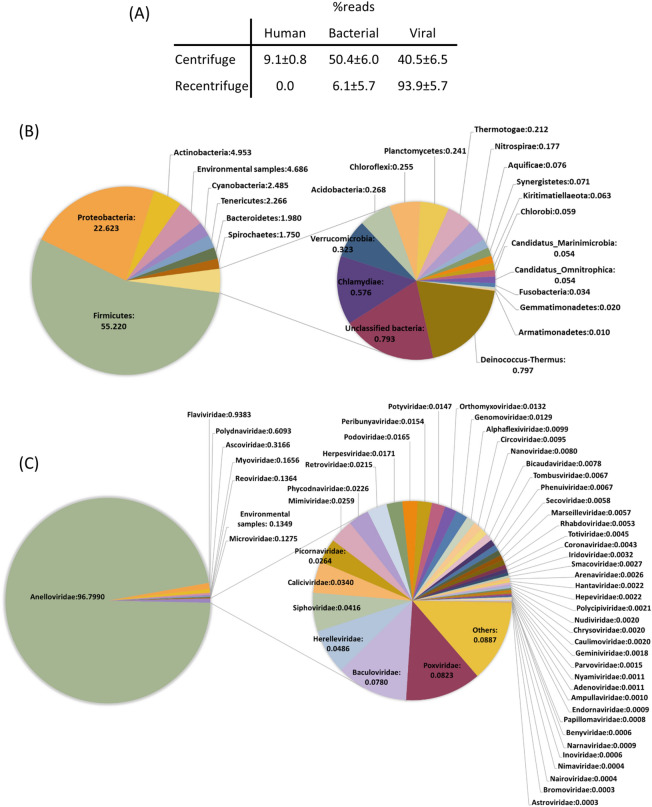
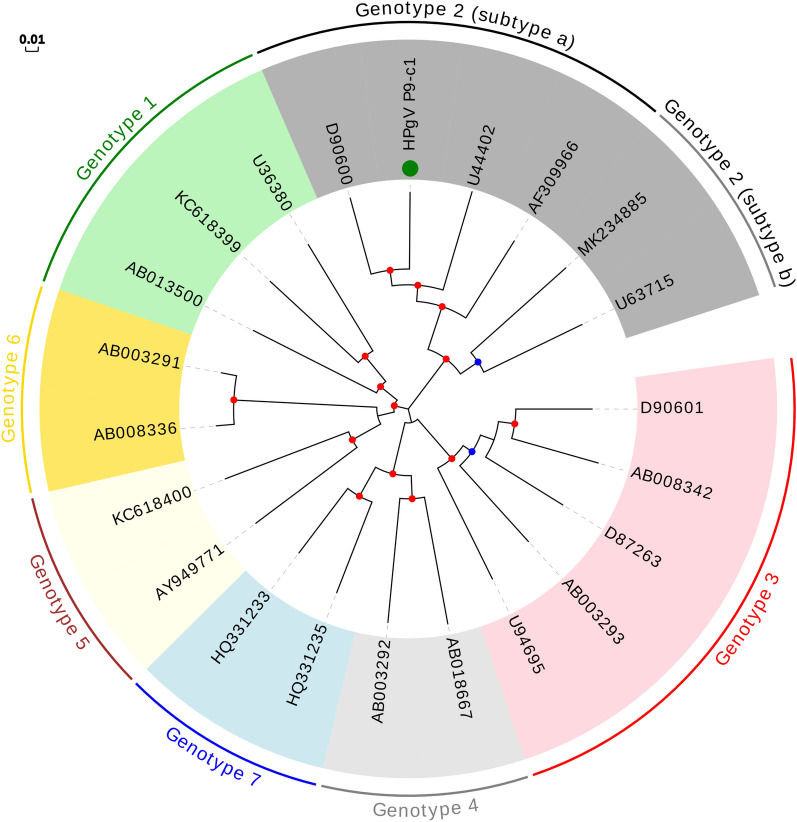
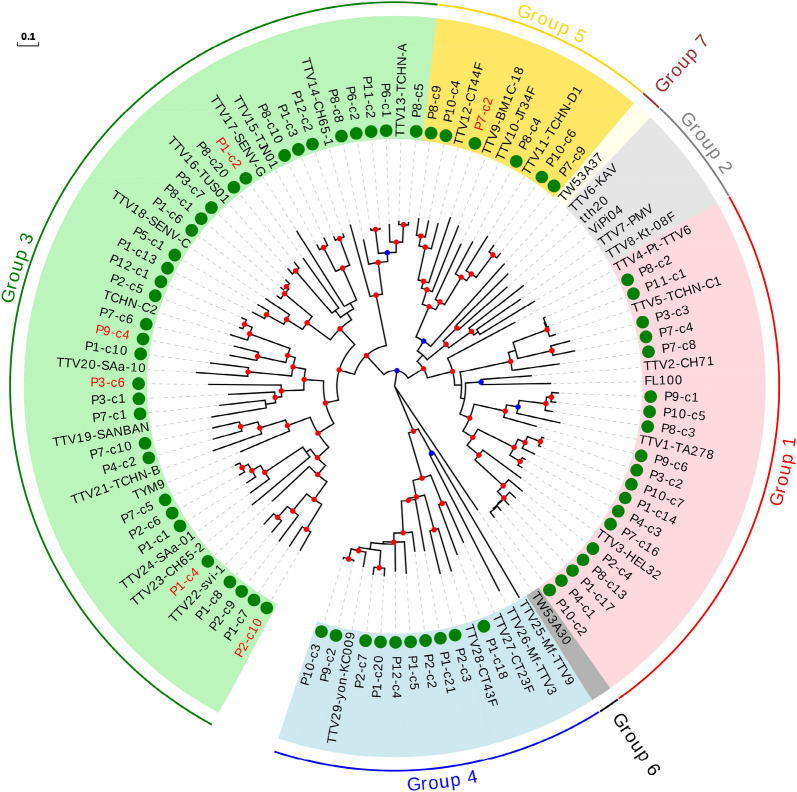
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
