

Mitochondrial aldehyde dehydrogenase (ALDH2) rescues cardiac contractile dysfunction in an APP/PS1 murine model of Alzheimer's disease via inhibition of ACSL4-dependent ferroptosis
- PMID: 33767380
- PMCID: PMC8724276
- DOI: 10.1038/s41401-021-00635-2
Mitochondrial aldehyde dehydrogenase (ALDH2) rescues cardiac contractile dysfunction in an APP/PS1 murine model of Alzheimer's disease via inhibition of ACSL4-dependent ferroptosis
Abstract
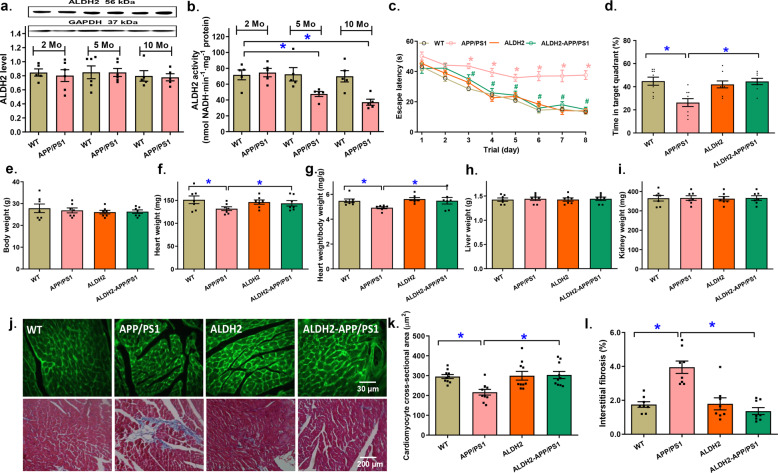
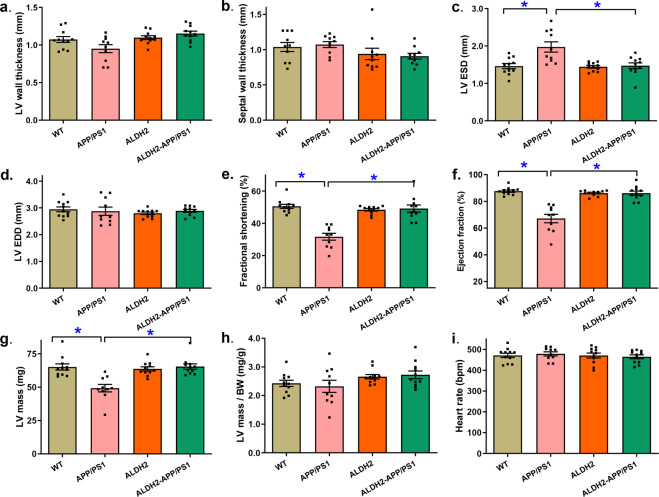
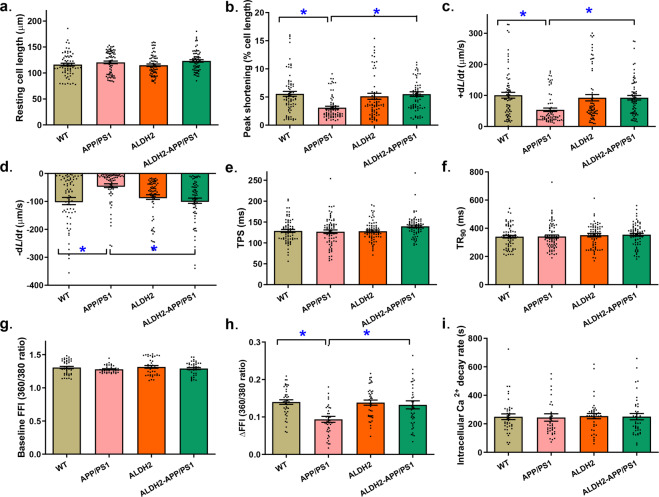
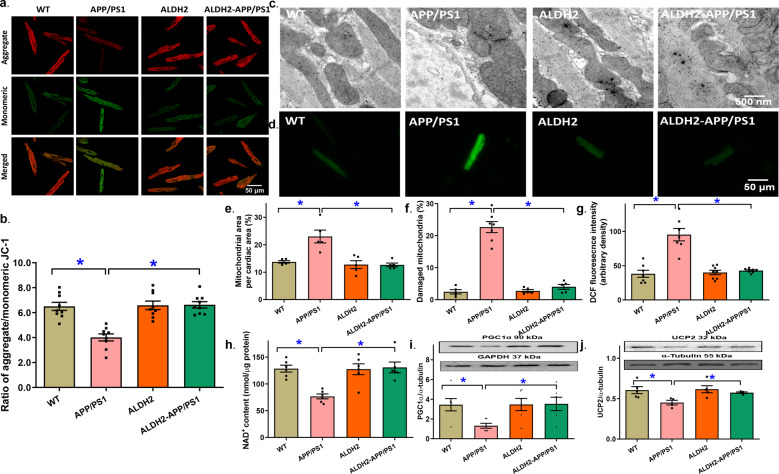
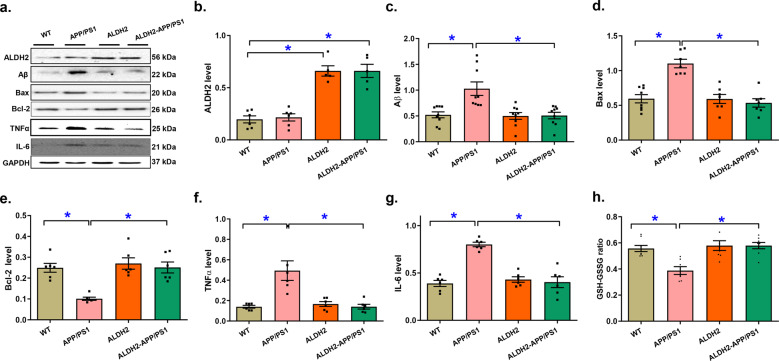
Alzheimer's disease (AD) is associated with high incidence of cardiovascular events but the mechanism remains elusive. Our previous study reveals a tight correlation between cardiac dysfunction and low mitochondrial aldehyde dehydrogenase (ALDH2) activity in elderly AD patients. In the present study we investigated the effect of ALDH2 overexpression on cardiac function in APP/PS1 mouse model of AD. Global ALDH2 transgenic mice were crossed with APP/PS1 mutant mice to generate the ALDH2-APP/PS1 mutant mice. Cognitive function, cardiac contractile, and morphological properties were assessed. We showed that APP/PS1 mice displayed significant cognitive deficit in Morris water maze test, myocardial ultrastructural, geometric (cardiac atrophy, interstitial fibrosis) and functional (reduced fractional shortening and cardiomyocyte contraction) anomalies along with oxidative stress, apoptosis, and inflammation in myocardium. ALDH2 transgene significantly attenuated or mitigated these anomalies. We also noted the markedly elevated levels of lipid peroxidation, the essential lipid peroxidation enzyme acyl-CoA synthetase long-chain family member 4 (ACSL4), the transcriptional regulator for ACLS4 special protein 1 (SP1) and ferroptosis, evidenced by elevated NCOA4, decreased GPx4, and SLC7A11 in myocardium of APP/PS1 mutant mice; these effects were nullified by ALDH2 transgene. In cardiomyocytes isolated from WT mice and in H9C2 myoblasts in vitro, application of Aβ (20 μM) decreased cell survival, compromised cardiomyocyte contractile function, and induced lipid peroxidation; ALDH2 transgene or activator Alda-1 rescued Aβ-induced deteriorating effects. ALDH2-induced protection against Aβ-induced lipid peroxidation was mimicked by the SP1 inhibitor tolfenamic acid (TA) or the ACSL4 inhibitor triacsin C (TC), and mitigated by the lipid peroxidation inducer 5-hydroxyeicosatetraenoic acid (5-HETE) or the ferroptosis inducer erastin. These results demonstrate an essential role for ALDH2 in AD-induced cardiac anomalies through regulation of lipid peroxidation and ferroptosis.
Keywords: ALDH2; Alda-1; Alzheimer’s disease; cardiac function; ferroptosis; landscape perceptions; lipid peroxidation; tolfenamic acid; triacsin C; 5-HETE; erastin.
© 2021. The Author(s), under exclusive licence to CPS and SIMM.
Conflict of interest statement
The authors declare no competing interests.
Figures



Similar articles
-
ALDH2 contributes to melatonin-induced protection against APP/PS1 mutation-prompted cardiac anomalies through cGAS-STING-TBK1-mediated regulation of mitophagy.Signal Transduct Target Ther. 2020 Jul 24;5(1):119. doi: 10.1038/s41392-020-0171-5. Signal Transduct Target Ther. 2020. PMID: 32703954 Free PMC article.
-
Impact of mitochondrial aldehyde dehydrogenase 2 on cognitive impairment in the AD model mouse.Acta Biochim Biophys Sin (Shanghai). 2021 Jul 5;53(7):837-847. doi: 10.1093/abbs/gmab057. Acta Biochim Biophys Sin (Shanghai). 2021. PMID: 33954430
-
Tetrahydroxy stilbene glycoside ameliorates Alzheimer's disease in APP/PS1 mice via glutathione peroxidase related ferroptosis.Int Immunopharmacol. 2021 Oct;99:108002. doi: 10.1016/j.intimp.2021.108002. Epub 2021 Jul 29. Int Immunopharmacol. 2021. PMID: 34333354
-
Mechanisms and preventive measures of ALDH2 in ischemia‑reperfusion injury: Ferroptosis as a novel target (Review).Mol Med Rep. 2025 Apr;31(4):105. doi: 10.3892/mmr.2025.13470. Epub 2025 Feb 28. Mol Med Rep. 2025. PMID: 40017132 Free PMC article. Review.
-
Acyl-CoA synthase ACSL4: an essential target in ferroptosis and fatty acid metabolism.Chin Med J (Engl). 2023 Nov 5;136(21):2521-2537. doi: 10.1097/CM9.0000000000002533. Chin Med J (Engl). 2023. PMID: 37442770 Free PMC article. Review.
Cited by
-
Vascular calcification: Molecular mechanisms and therapeutic interventions.MedComm (2020). 2023 Jan 3;4(1):e200. doi: 10.1002/mco2.200. eCollection 2023 Feb. MedComm (2020). 2023. PMID: 36620697 Free PMC article. Review.
-
Circular RNA 0016142 Knockdown Induces Ferroptosis in Hepatocellular Carcinoma Cells via Modulation of the MicroRNA-188-3p/Glutathione Peroxidase 4 Axis.Biochem Genet. 2024 Feb;62(1):333-351. doi: 10.1007/s10528-023-10417-6. Epub 2023 Jun 21. Biochem Genet. 2024. PMID: 37344692
-
Oral administration of butylated hydroxytoluene induces neuroprotection in a streptozotocin-induced rat Alzheimer's disease model via inhibition of neuronal ferroptosis.Mol Med. 2024 Nov 8;30(1):204. doi: 10.1186/s10020-024-00980-y. Mol Med. 2024. PMID: 39511487 Free PMC article.
-
The Interplay Between Autophagy and Regulated Necrosis.Antioxid Redox Signal. 2023 Mar;38(7-9):550-580. doi: 10.1089/ars.2022.0110. Epub 2022 Oct 12. Antioxid Redox Signal. 2023. PMID: 36053716 Free PMC article. Review.
-
Impact of common ALDH2 inactivating mutation and alcohol consumption on Alzheimer's disease.Front Aging Neurosci. 2023 Aug 24;15:1223977. doi: 10.3389/fnagi.2023.1223977. eCollection 2023. Front Aging Neurosci. 2023. PMID: 37693648 Free PMC article. Review.
References
-
- Jellinger KA. Pathobiological subtypes of Alzheimer disease. Dement Geriatr Cogn Disord. 2020;49:321–33. - PubMed
-
- Johnson J, Mercado-Ayon E, Mercado-Ayon Y, Dong YN, Halawani S, Ngaba L, et al. Mitochondrial dysfunction in the development and progression of neurodegenerative diseases. Arch Biochem Biophys 2021. 10.1016/j.abb.2020.108698. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
