

Ultra-fast proteomics with Scanning SWATH
- PMID: 33767396
- PMCID: PMC7611254
- DOI: 10.1038/s41587-021-00860-4
Ultra-fast proteomics with Scanning SWATH
Abstract
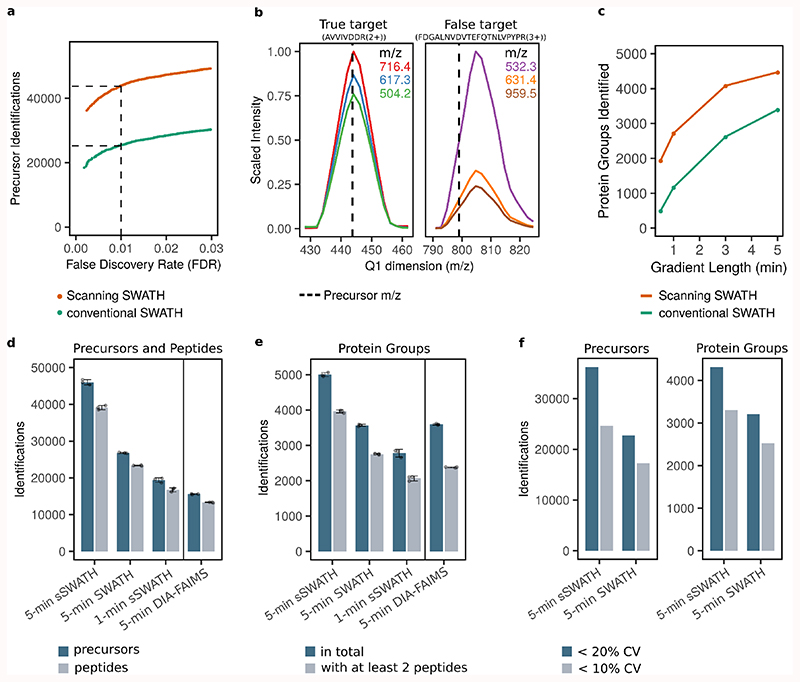
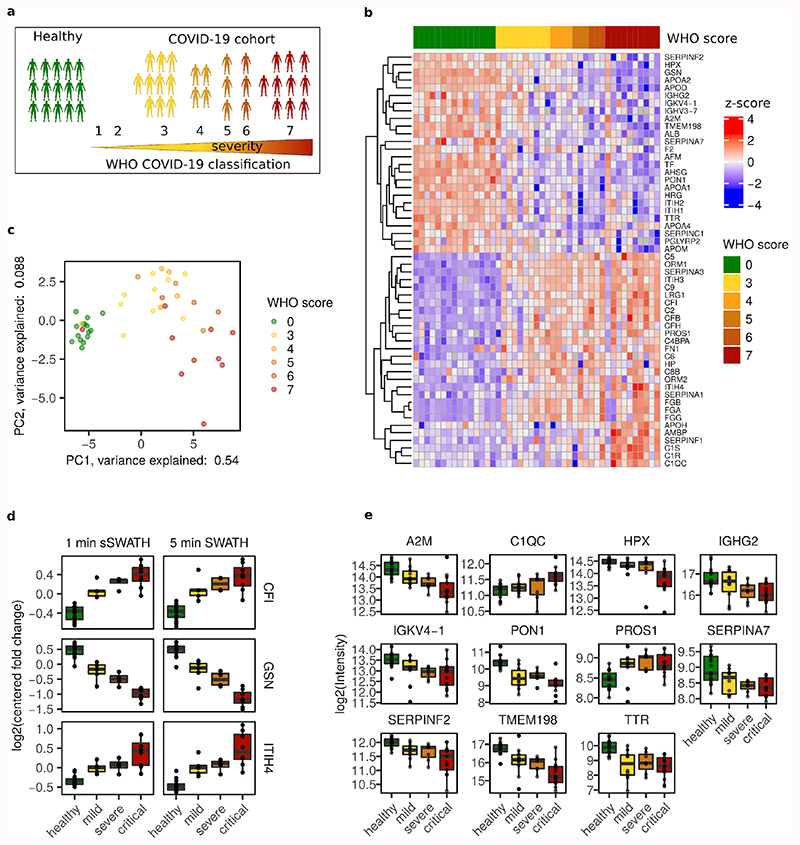
Accurate quantification of the proteome remains challenging for large sample series and longitudinal experiments. We report a data-independent acquisition method, Scanning SWATH, that accelerates mass spectrometric (MS) duty cycles, yielding quantitative proteomes in combination with short gradients and high-flow (800 µl min-1) chromatography. Exploiting a continuous movement of the precursor isolation window to assign precursor masses to tandem mass spectrometry (MS/MS) fragment traces, Scanning SWATH increases precursor identifications by ~70% compared to conventional data-independent acquisition (DIA) methods on 0.5-5-min chromatographic gradients. We demonstrate the application of ultra-fast proteomics in drug mode-of-action screening and plasma proteomics. Scanning SWATH proteomes capture the mode of action of fungistatic azoles and statins. Moreover, we confirm 43 and identify 11 new plasma proteome biomarkers of COVID-19 severity, advancing patient classification and biomarker discovery. Thus, our results demonstrate a substantial acceleration and increased depth in fast proteomic experiments that facilitate proteomic drug screens and clinical studies.
© 2021. The Author(s), under exclusive licence to Springer Nature America, Inc. part of Springer Nature.
Conflict of interest statement
N.B, G.I., F.W and S.T. work for SCIEX. All other authors have no competing interests.
Figures


Comment in
-
Increasing proteomics throughput.Nat Biotechnol. 2021 Jul;39(7):809-810. doi: 10.1038/s41587-021-00881-z. Nat Biotechnol. 2021. PMID: 33767394 Free PMC article. No abstract available.
References
-
- Aebersold R, Mann M. Mass-spectrometric exploration of proteome structure and function. Nature. 2016;537:347–355. - PubMed
-
- Cox J, Mann M. Quantitative, high-resolution proteomics for data-driven systems biology. Annu Rev Biochem. 2011;80:273–299. - PubMed
-
- Nilsson T, et al. Mass spectrometry in high-throughput proteomics: ready for the big time. Nat Methods. 2010;7:681–685. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
