

A first exon termination checkpoint preferentially suppresses extragenic transcription
- PMID: 33767452
- PMCID: PMC7610630
- DOI: 10.1038/s41594-021-00572-y
A first exon termination checkpoint preferentially suppresses extragenic transcription
Abstract
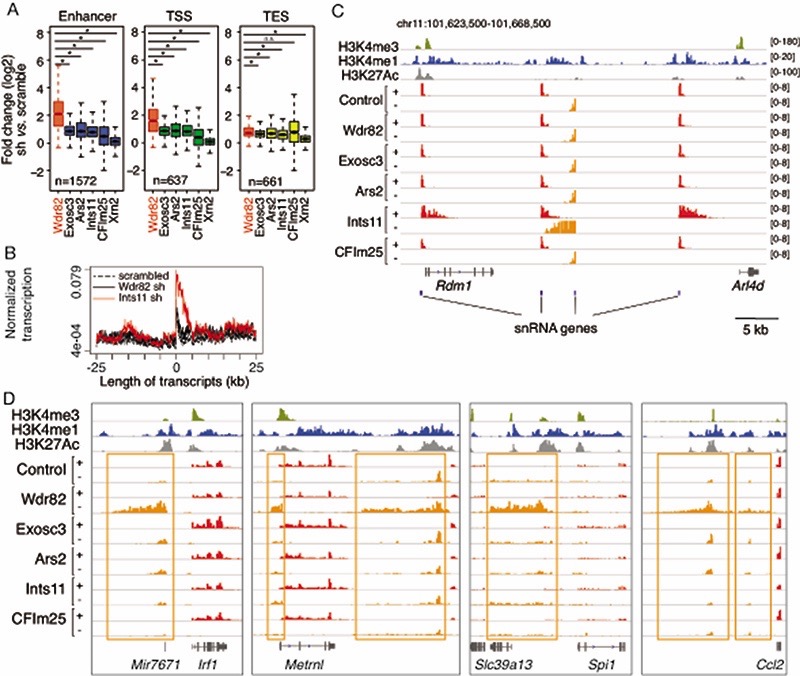
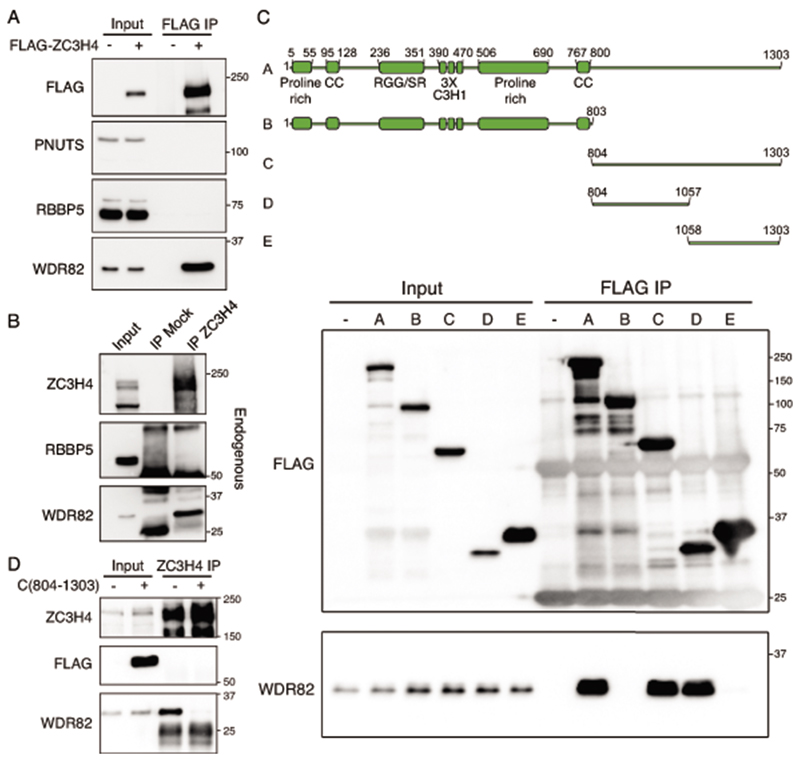
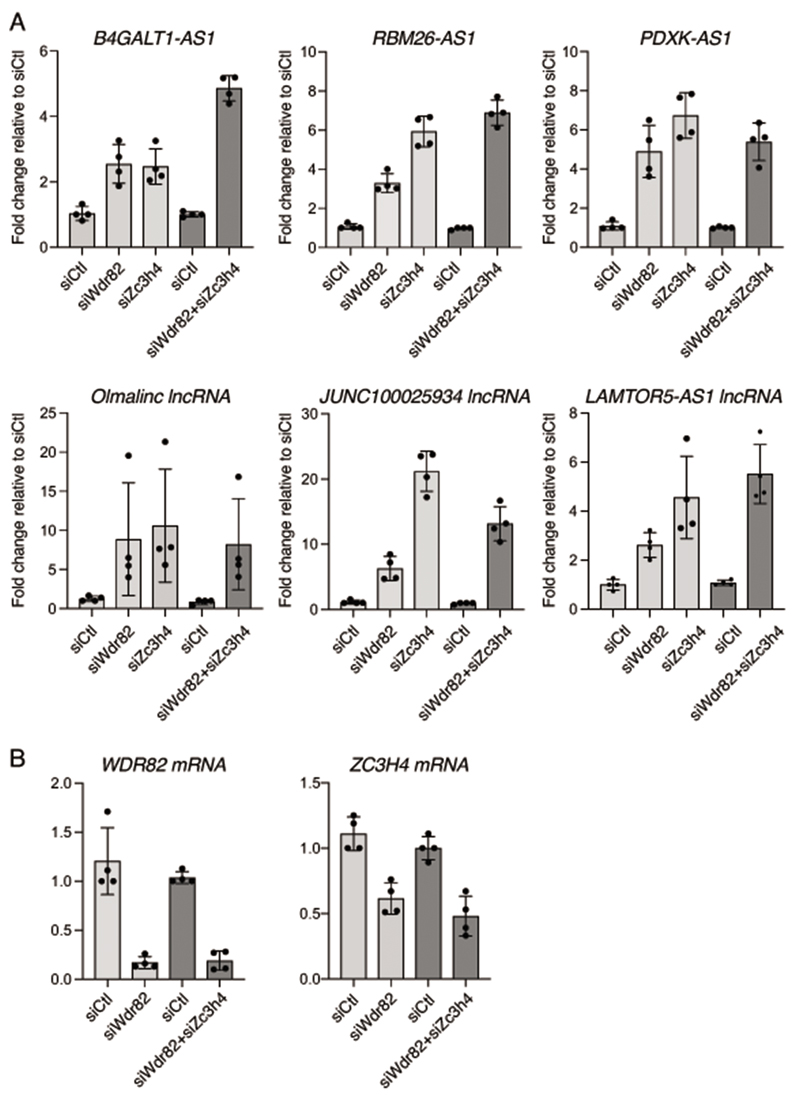
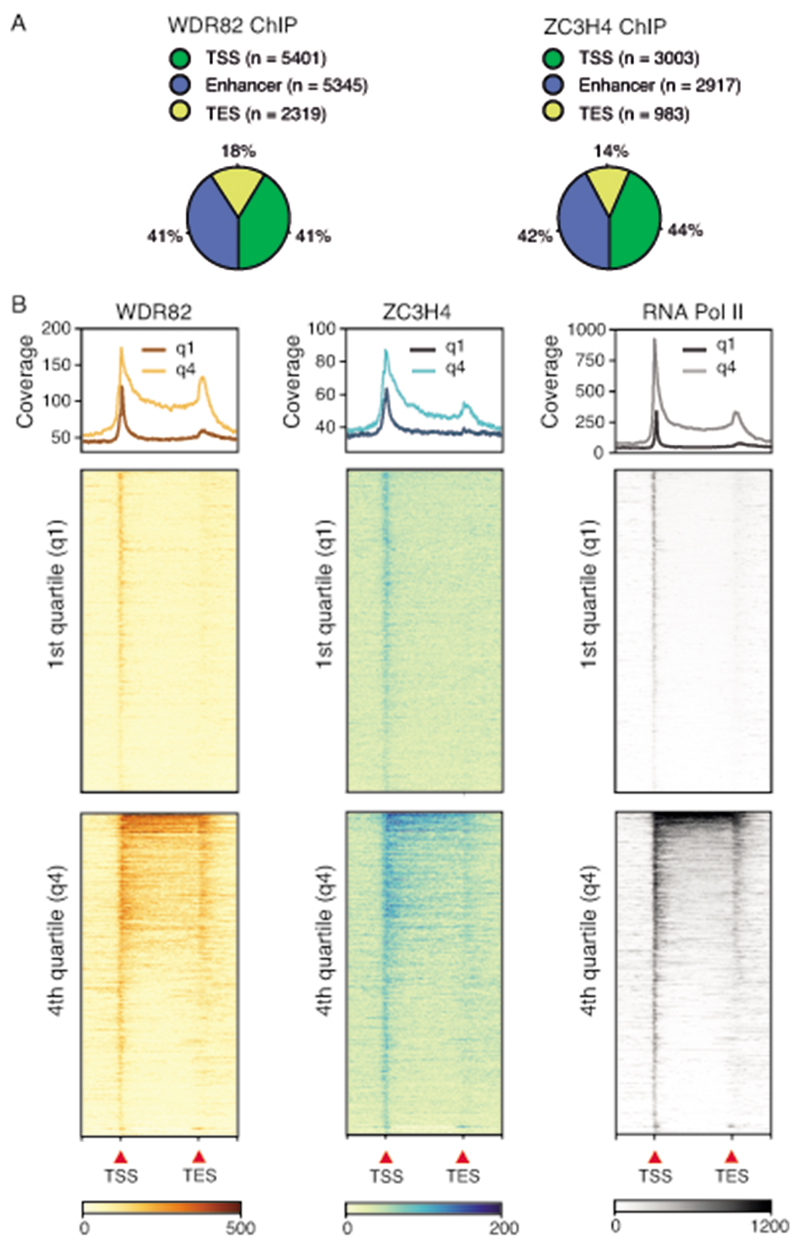
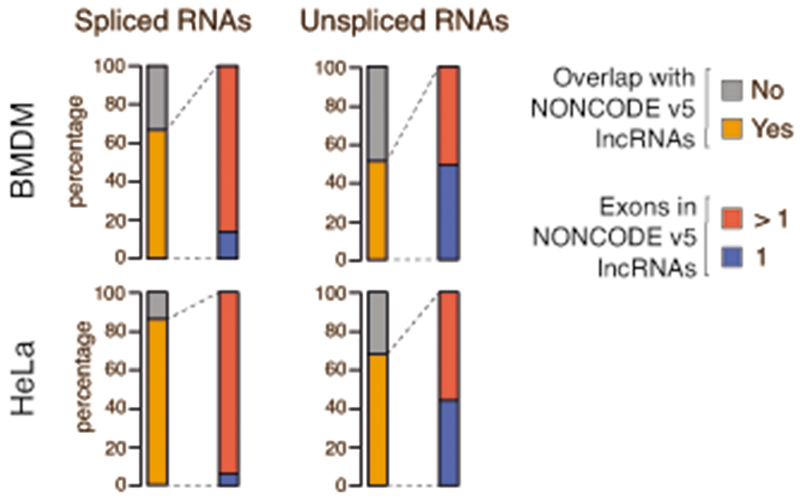
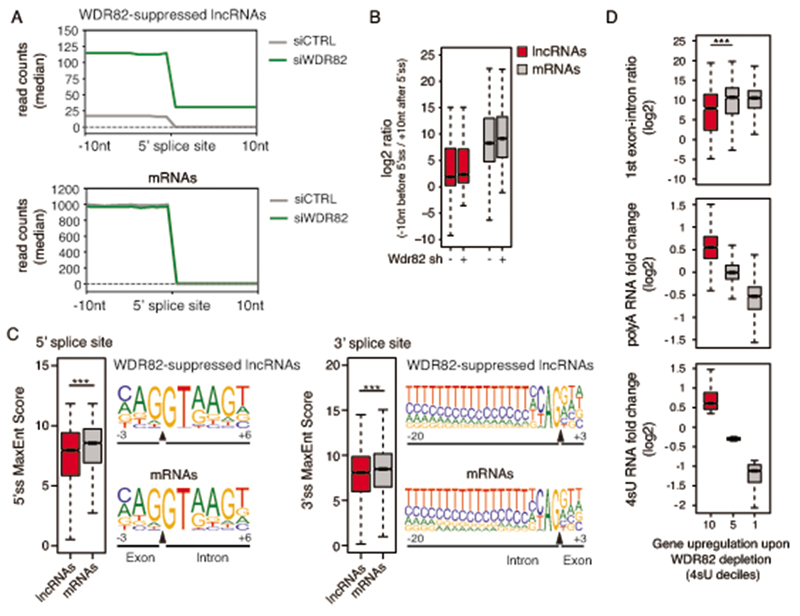
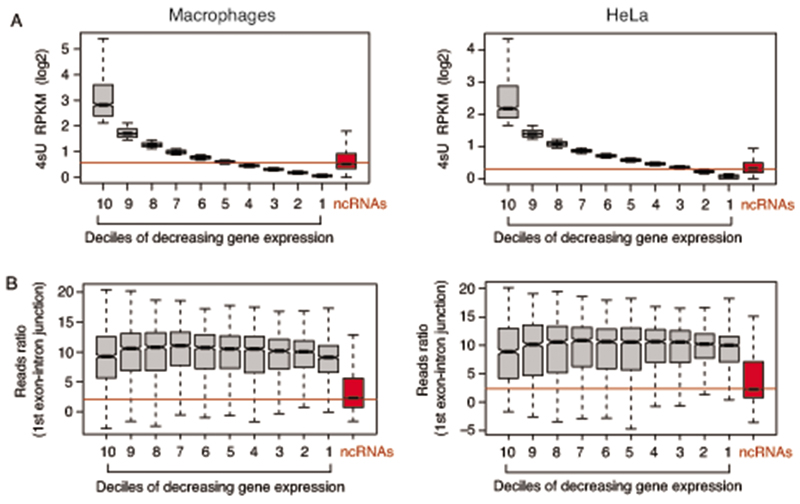
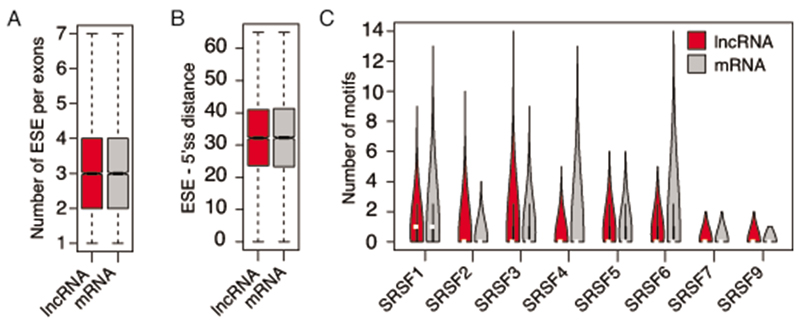
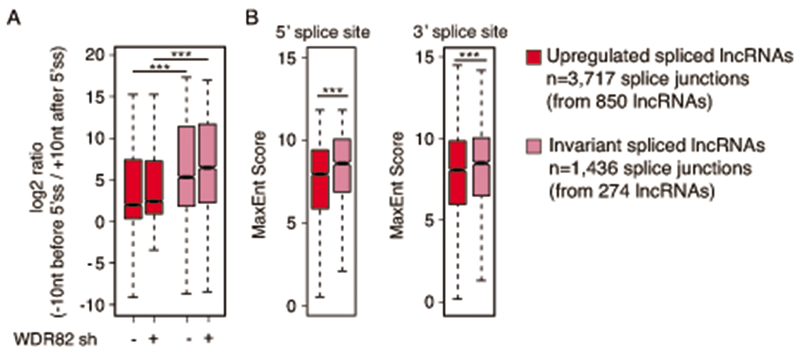
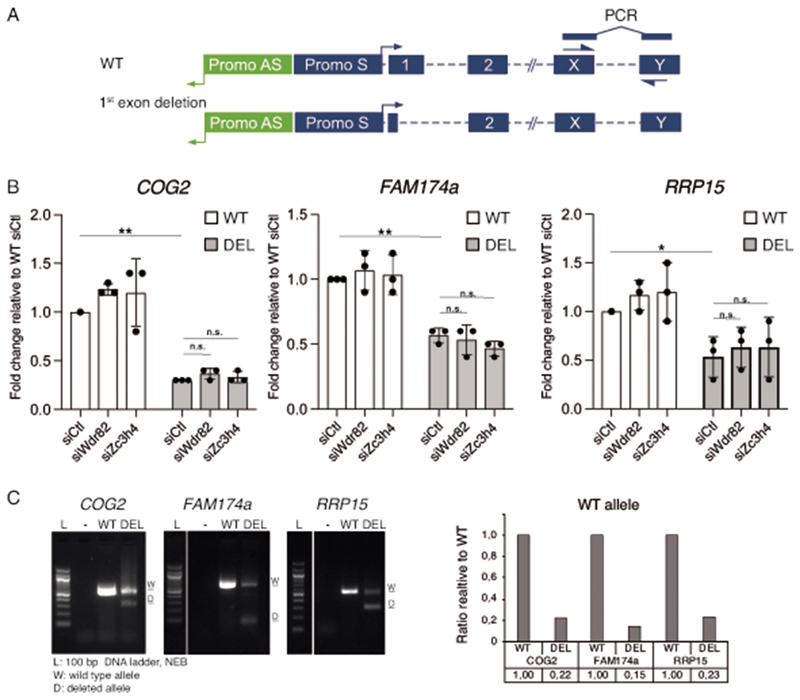
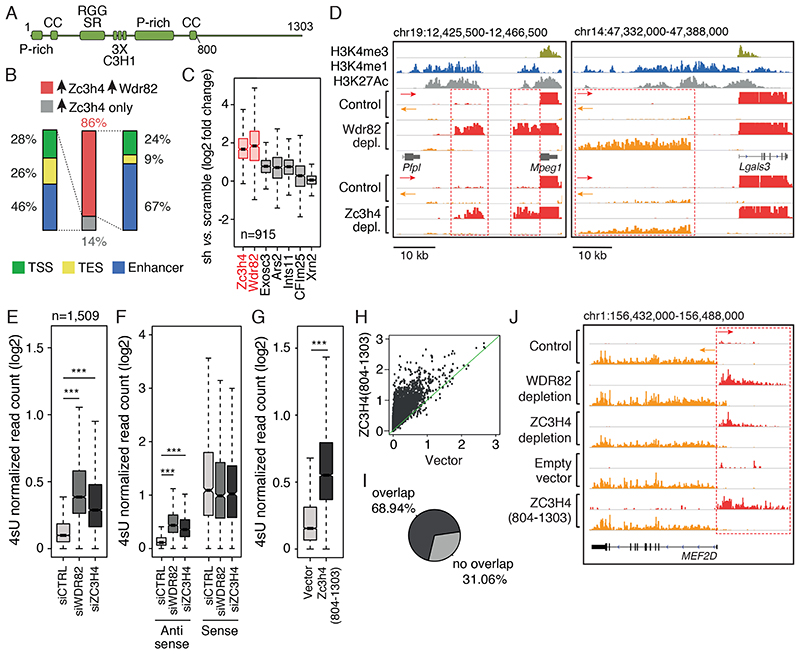
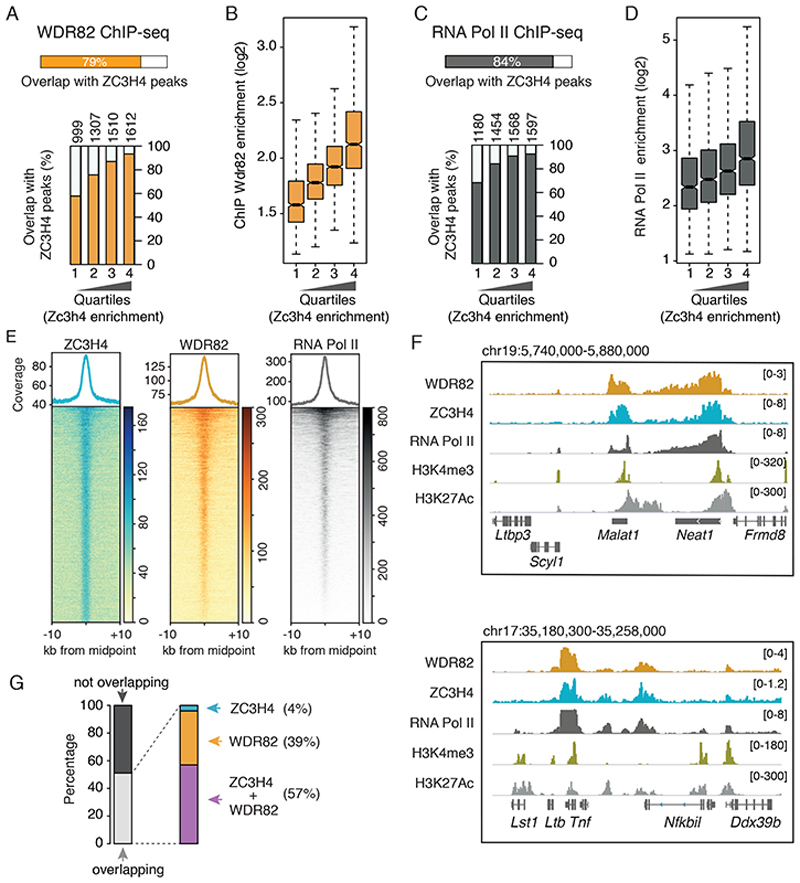
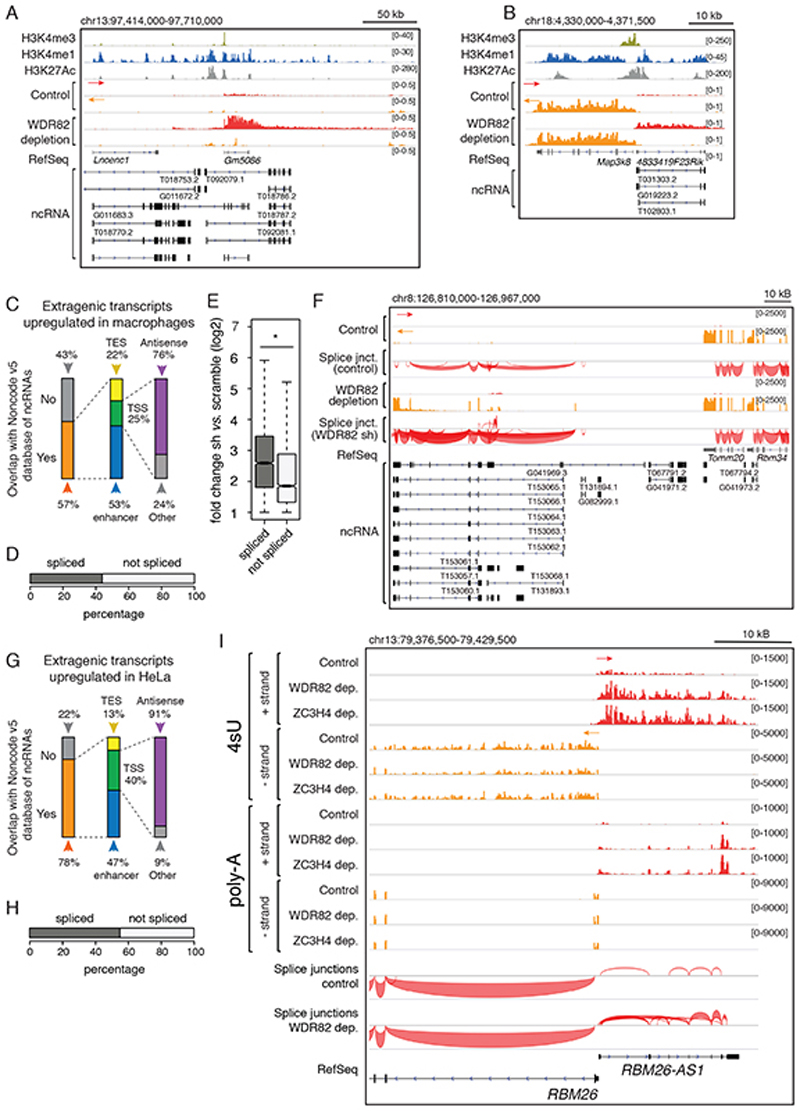
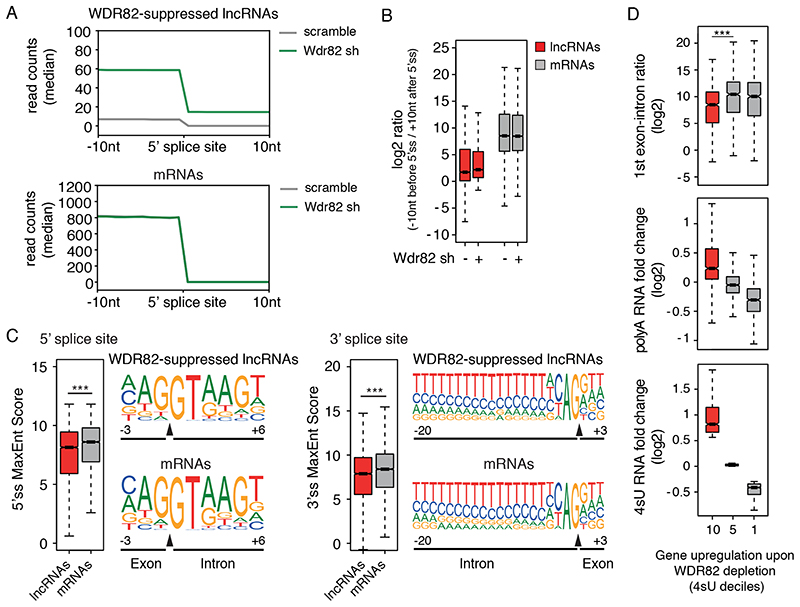
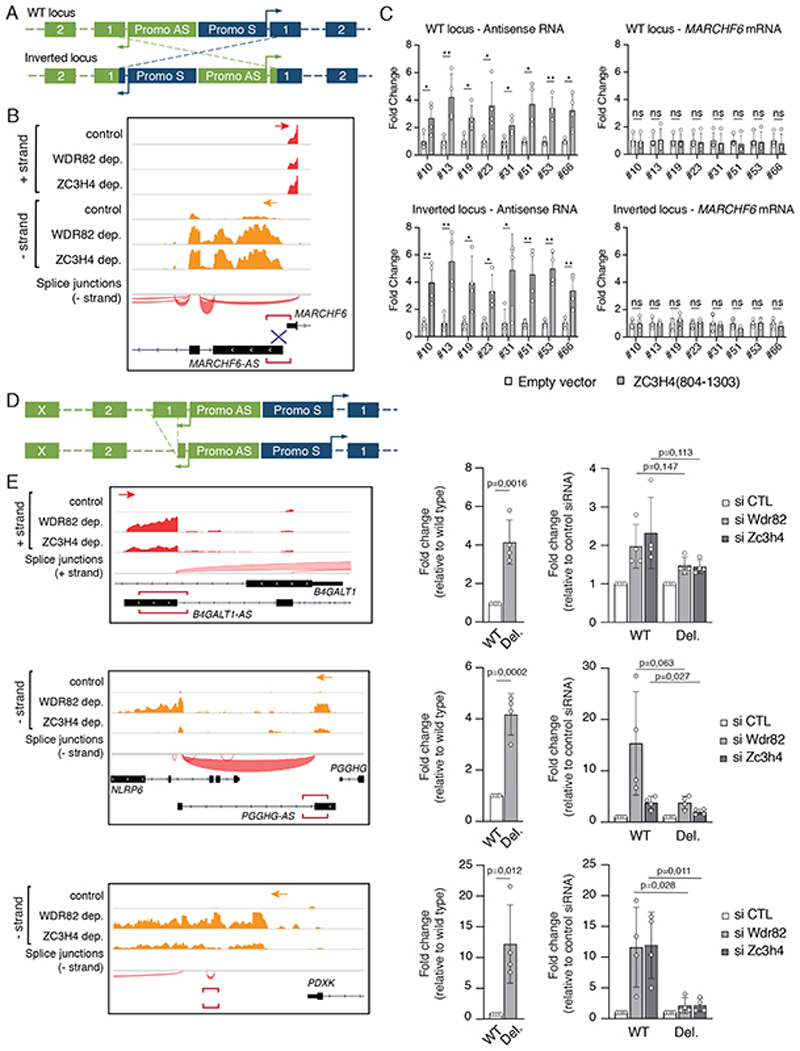
Interactions between the splicing machinery and RNA polymerase II increase protein-coding gene transcription. Similarly, exons and splicing signals of enhancer-generated long noncoding RNAs (elncRNAs) augment enhancer activity. However, elncRNAs are inefficiently spliced, suggesting that, compared with protein-coding genes, they contain qualitatively different exons with a limited ability to drive splicing. We show here that the inefficiently spliced first exons of elncRNAs as well as promoter-antisense long noncoding RNAs (pa-lncRNAs) in human and mouse cells trigger a transcription termination checkpoint that requires WDR82, an RNA polymerase II-binding protein, and its RNA-binding partner of previously unknown function, ZC3H4. We propose that the first exons of elncRNAs and pa-lncRNAs are an intrinsic component of a regulatory mechanism that, on the one hand, maximizes the activity of these cis-regulatory elements by recruiting the splicing machinery and, on the other, contains elements that suppress pervasive extragenic transcription.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
