

A Novel Pathogenic HSPG2 Mutation in Schwartz-Jampel Syndrome
- PMID: 33767660
- PMCID: PMC7985266
- DOI: 10.3389/fneur.2021.632336
A Novel Pathogenic HSPG2 Mutation in Schwartz-Jampel Syndrome
Abstract
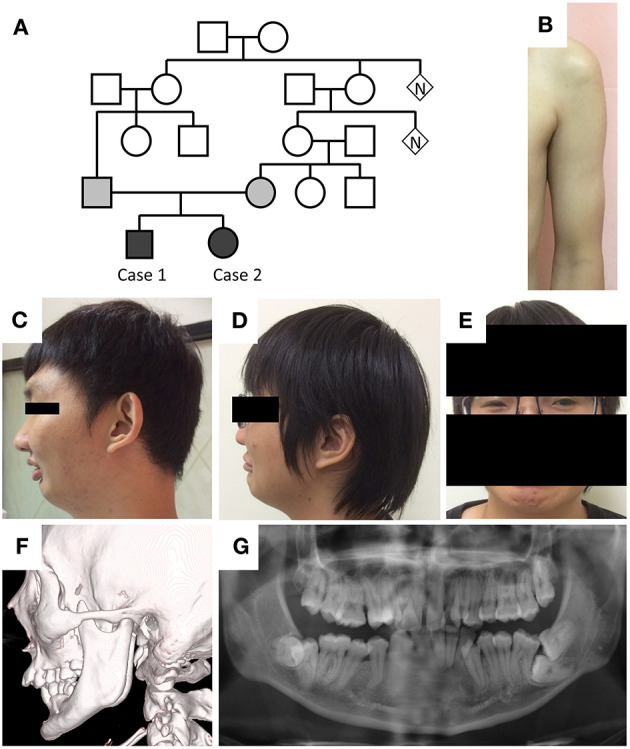
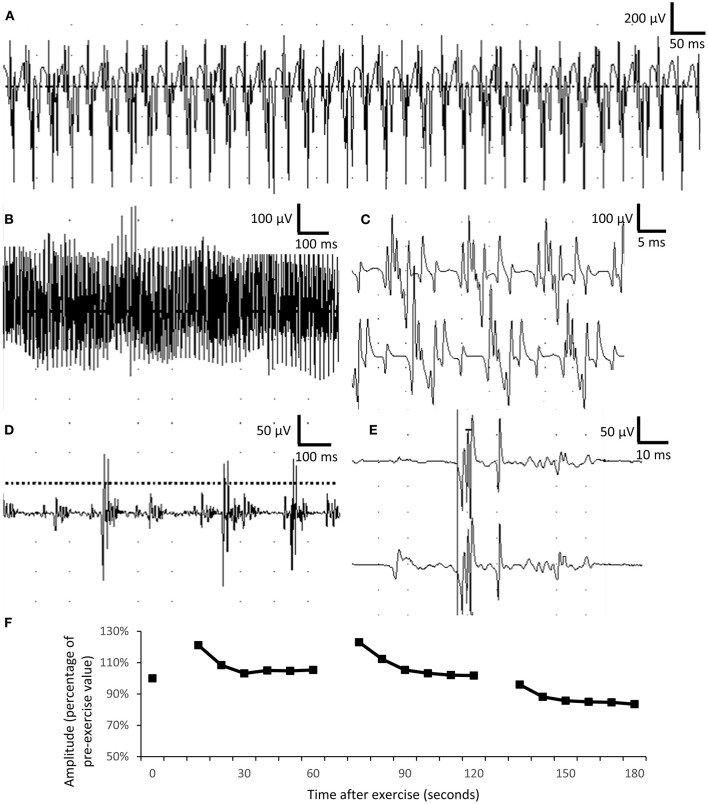
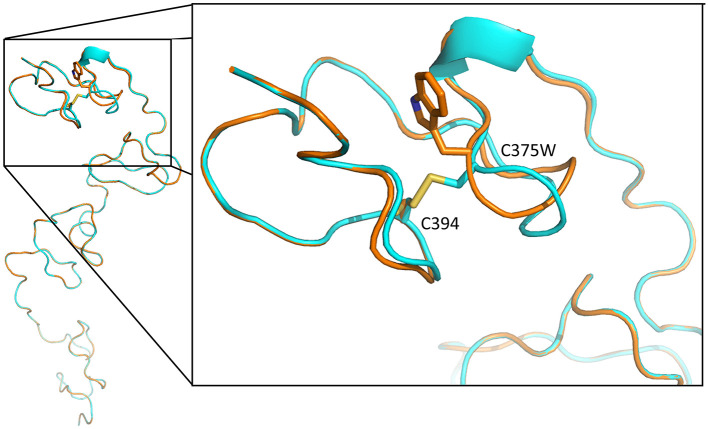
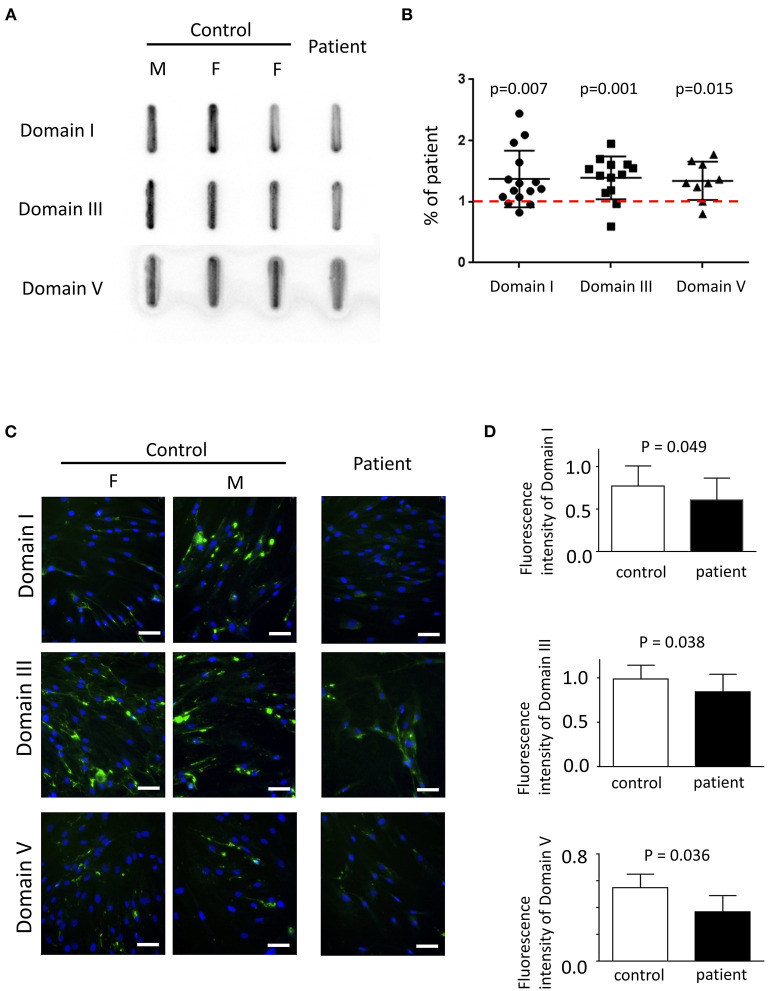
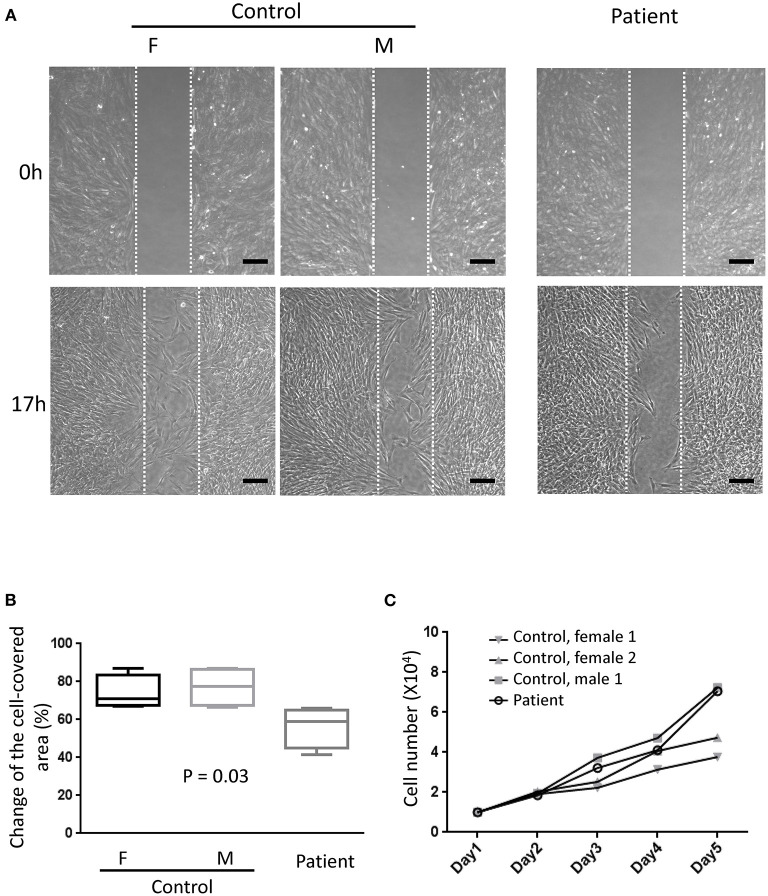
Schwartz-Jampel syndrome is a rare autosomal recessive disease caused by mutation in the heparan sulfate proteoglycan 2 (HSPG2) gene. Its cardinal symptoms are skeletal dysplasia and neuromuscular hyperactivity. Herein, we identified a new pathogenic mutation site (NM_005529.6:c.1125C>G; p.Cys375Trp) of HSPG2 leading to Schwartz-Jampel syndrome by whole-exome sequencing. This mutation carried by the asymptomatic parents was previously registered in a single-nucleotide polymorphism database of the National Institutes of Health as a coding sequence variant rs543805444. The pathogenic nature of this missense mutation was demonstrated by in silico pathogenicity assessment, clinical presentations, and cellular function of primary fibroblast derived from patients. Various in silico software applications predicted the mutation to be pathogenic [Sorting Intolerant From Tolerant (SIFT), 0; Polyphen-2, 1; CADD (Combined Annotation Dependent Depletion), 23.7; MutationTaster, 1; DANN (deleterious annotation of genetic variants using neural networks); 0.9]. Needle electromyography revealed extensive complex repetitive discharges and multiple polyphasic motor unit action potentials in axial and limb muscles at rest. Short exercise test for myotonia showed Fournier pattern I. At cellular levels, mutant primary fibroblasts had reduced levels of secreted perlecan and impaired migration ability but normal capability of proliferation. Patients with this mutation showed more neuromuscular instability and relatively mild skeletal abnormality comparing with previously reported cases.
Keywords: Schwartz–Jampel syndrome; heparan sulfate proteoglycan 2; perlecan; primary fibroblast; short exercise test.
Copyright © 2021 Lin, Hung, Hsu, Chang and Sun.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources