

Identification and Analysis of Potential Key Genes Associated With Hepatocellular Carcinoma Based on Integrated Bioinformatics Methods
- PMID: 33767726
- PMCID: PMC7985067
- DOI: 10.3389/fgene.2021.571231
Identification and Analysis of Potential Key Genes Associated With Hepatocellular Carcinoma Based on Integrated Bioinformatics Methods
Abstract
Background: Hepatocellular carcinoma (HCC) is a type of primary liver tumor with poor prognosis and high mortality, and its molecular mechanism remains incompletely understood. This study aimed to use bioinformatics technology to identify differentially expressed genes (DEGs) in HCC pathogenesis, hoping to identify novel biomarkers or potential therapeutic targets for HCC research.
Methods: The bioinformatics analysis of our research mostly involved the following two datasets: Gene Expression Omnibus (GEO) and The Cancer Genome Atlas (TCGA). First, we screened DEGs based on the R packages (limma and edgeR). Using the DAVID database, the Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses of DEGs were carried out. Next, the protein-protein interaction (PPI) network of the DEGs was built in the STRING database. Then, hub genes were screened through the cytoHubba plug-in, followed by verification using the GEPIA and Oncomine databases. We demonstrated differences in levels of the protein in hub genes using the Human Protein Atlas (HPA) database. Finally, the hub genes prognostic values were analyzed by the GEPIA database. Additionally, using the Comparative Toxicogenomics Database (CTD), we constructed the drug-gene interaction network.
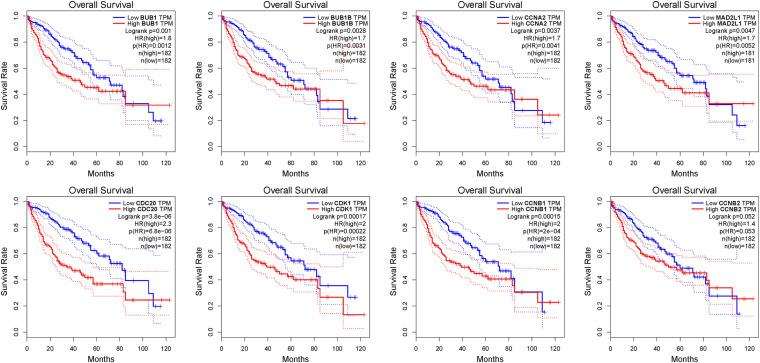
Results: We ended up with 763 DEGs, including 247 upregulated and 516 downregulated DEGs, that were mainly enriched in the epoxygenase P450 pathway, oxidation-reduction process, and metabolism-related pathways. Through the constructed PPI network, it can be concluded that the P53 signaling pathway and the cell cycle are the most obvious in module analysis. From the PPI, we filtered out eight hub genes, and these genes were significantly upregulated in HCC samples, findings consistent with the expression validation results. Additionally, survival analysis showed that high level gene expression of CDC20, CDK1, MAD2L1, BUB1, BUB1B, CCNB1, and CCNA2 were connected with the poor overall survival of HCC patients. Toxicogenomics analysis showed that only topotecan, oxaliplatin, and azathioprine could reduce the gene expression levels of all seven hub genes.
Conclusion: The present study screened out the key genes and pathways that were related to HCC pathogenesis, which could provide new insight for the future molecularly targeted therapy and prognosis evaluation of HCC.
Keywords: GEO; TCGA; bioinformatics; biomarker; differentially expressed genes; hepatocellular carcinoma; survival.
Copyright © 2021 Li, Lin, Cheng, Zhang and Cai.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures







References
-
- Aufhauser D. D., Jr., Sadot E., Murken D. R., Eddinger K., Hoteit M., Abt P. L., et al. (2018). Incidence of occult intrahepatic metastasis in hepatocellular carcinoma treated with transplantation corresponds to early recurrence rates after partial hepatectomy. Ann. Surg. 267 922–928. 10.1097/sla.0000000000002135 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous