

Kcnq2/Kv7.2 controls the threshold and bi-hemispheric symmetry of cortical spreading depolarization
- PMID: 33768249
- PMCID: PMC8536937
- DOI: 10.1093/brain/awab141
Kcnq2/Kv7.2 controls the threshold and bi-hemispheric symmetry of cortical spreading depolarization
Abstract
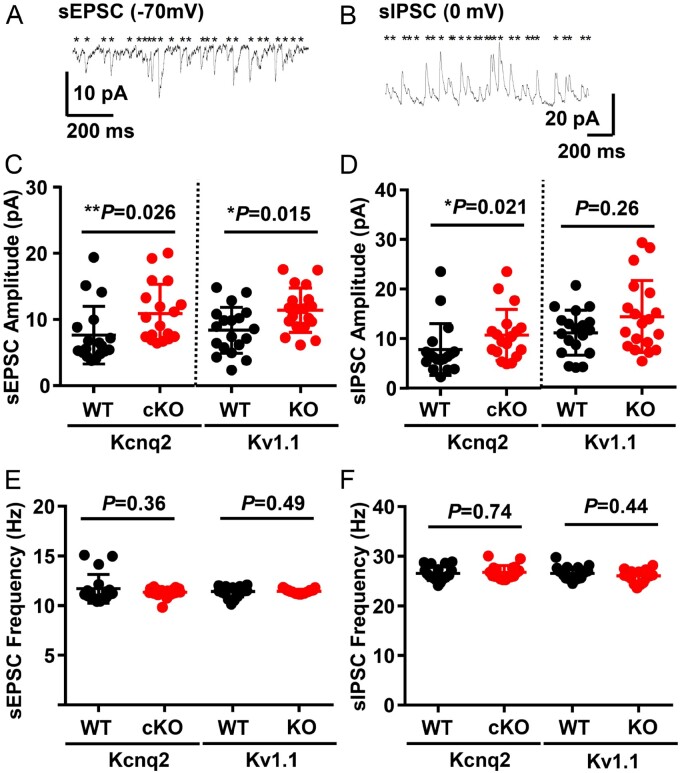
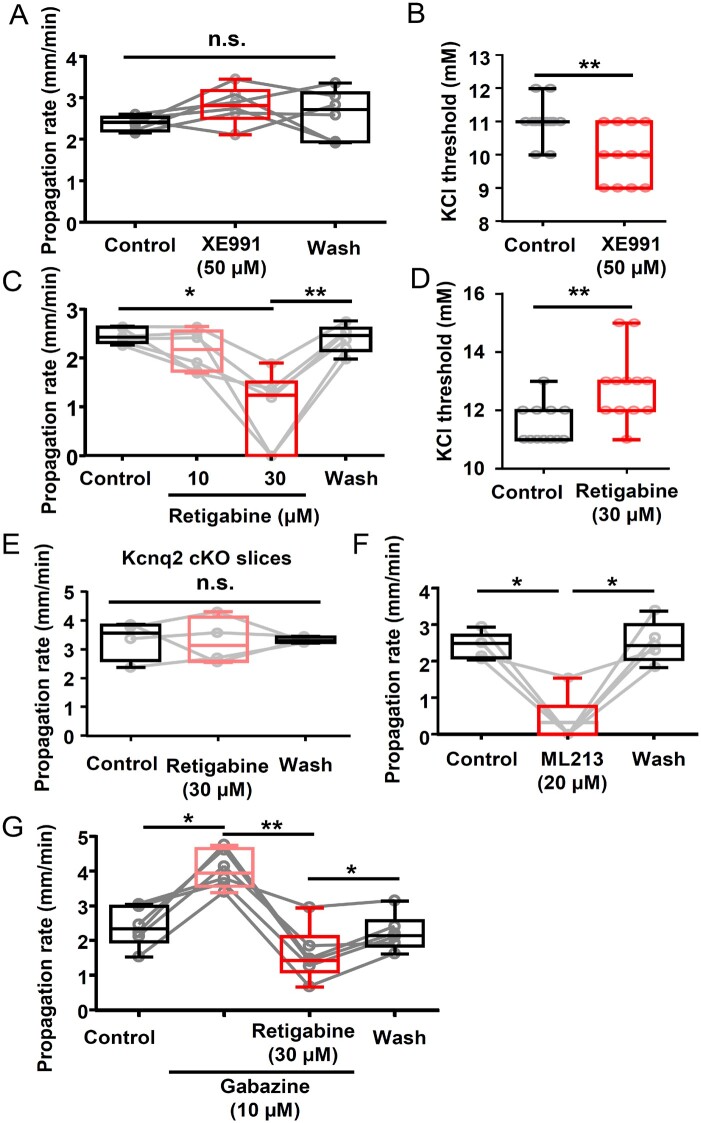
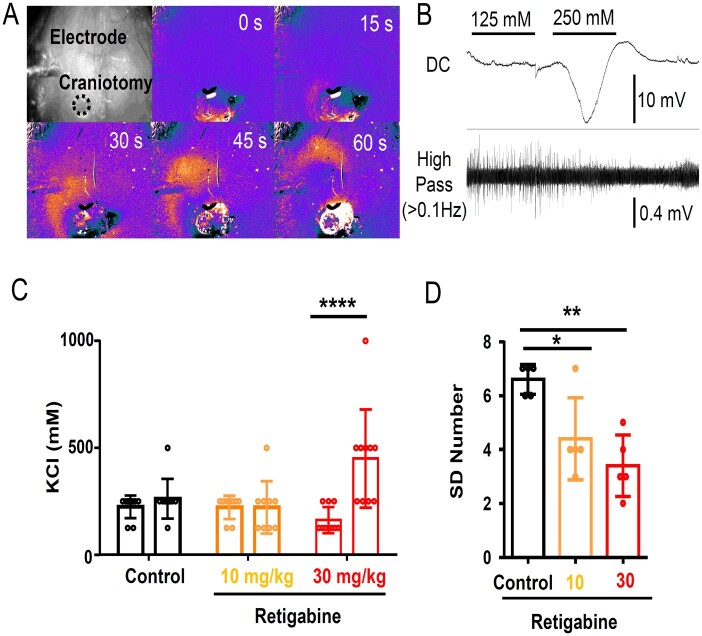
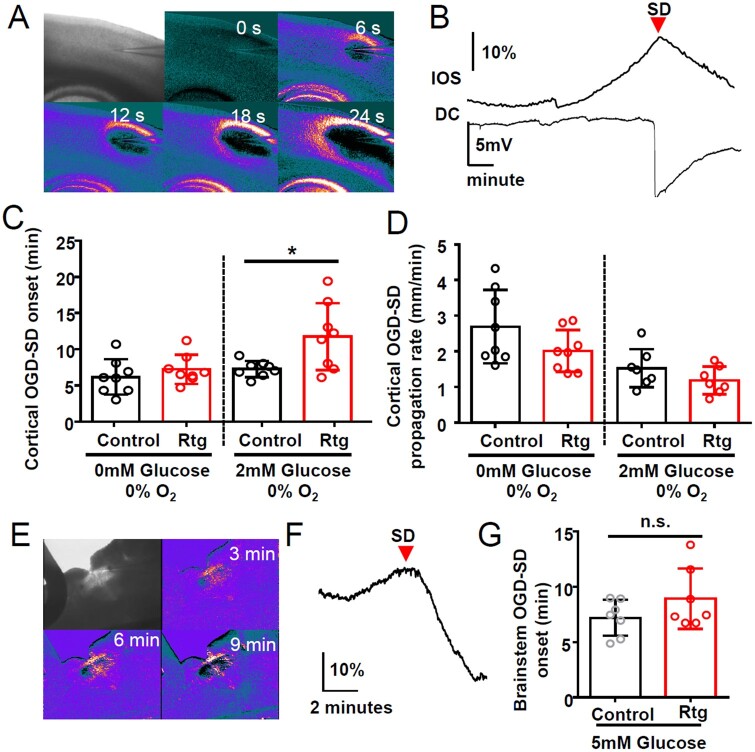
Spreading depolarization is a slowly propagating wave of massive cellular depolarization associated with acute brain injury and migraine aura. Genetic studies link depolarizing molecular defects in Ca2+ flux, Na+ current in interneurons, and glial Na+-K+ ATPase with spreading depolarization susceptibility, emphasizing the important roles of synaptic activity and extracellular ionic homeostasis in determining spreading depolarization threshold. In contrast, although gene mutations in voltage-gated potassium ion channels that shape intrinsic membrane excitability are frequently associated with epilepsy susceptibility, it is not known whether epileptogenic mutations that regulate membrane repolarization also modify spreading depolarization threshold and propagation. Here we report that the Kcnq2/Kv7.2 potassium channel subunit, frequently mutated in developmental epilepsy, is a spreading depolarization modulatory gene with significant control over the seizure-spreading depolarization transition threshold, bi-hemispheric cortical expression, and diurnal temporal susceptibility. Chronic DC-band cortical EEG recording from behaving conditional Kcnq2 deletion mice (Emx1cre/+::Kcnq2flox/flox) revealed spontaneous cortical seizures and spreading depolarization. In contrast to the related potassium channel deficient model, Kv1.1-KO mice, spontaneous cortical spreading depolarizations in Kcnq2 cKO mice are tightly coupled to the terminal phase of seizures, arise bilaterally, and are observed predominantly during the dark phase. Administration of the non-selective Kv7.2 inhibitor XE991 to Kv1.1-KO mice partly reproduced the Kcnq2 cKO-like spreading depolarization phenotype (tight seizure coupling and bilateral symmetry) in these mice, indicating that Kv7.2 currents can directly and actively modulate spreading depolarization properties. In vitro brain slice studies confirmed that Kcnq2/Kv7.2 depletion or pharmacological inhibition intrinsically lowers the cortical spreading depolarization threshold, whereas pharmacological Kv7.2 activators elevate the threshold to multiple depolarizing and hypometabolic spreading depolarization triggers. Together these results identify Kcnq2/Kv7.2 as a distinctive spreading depolarization regulatory gene, and point to spreading depolarization as a potentially significant pathophysiological component of KCNQ2-linked epileptic encephalopathy syndromes. Our results also implicate KCNQ2/Kv7.2 channel activation as a potential adjunctive therapeutic target to inhibit spreading depolarization incidence.
Keywords: Kv1.1; XE991; epilepsy; retigabine; seizure.
© The Author(s) (2021). Published by Oxford University Press on behalf of the Guarantors of Brain.
Figures
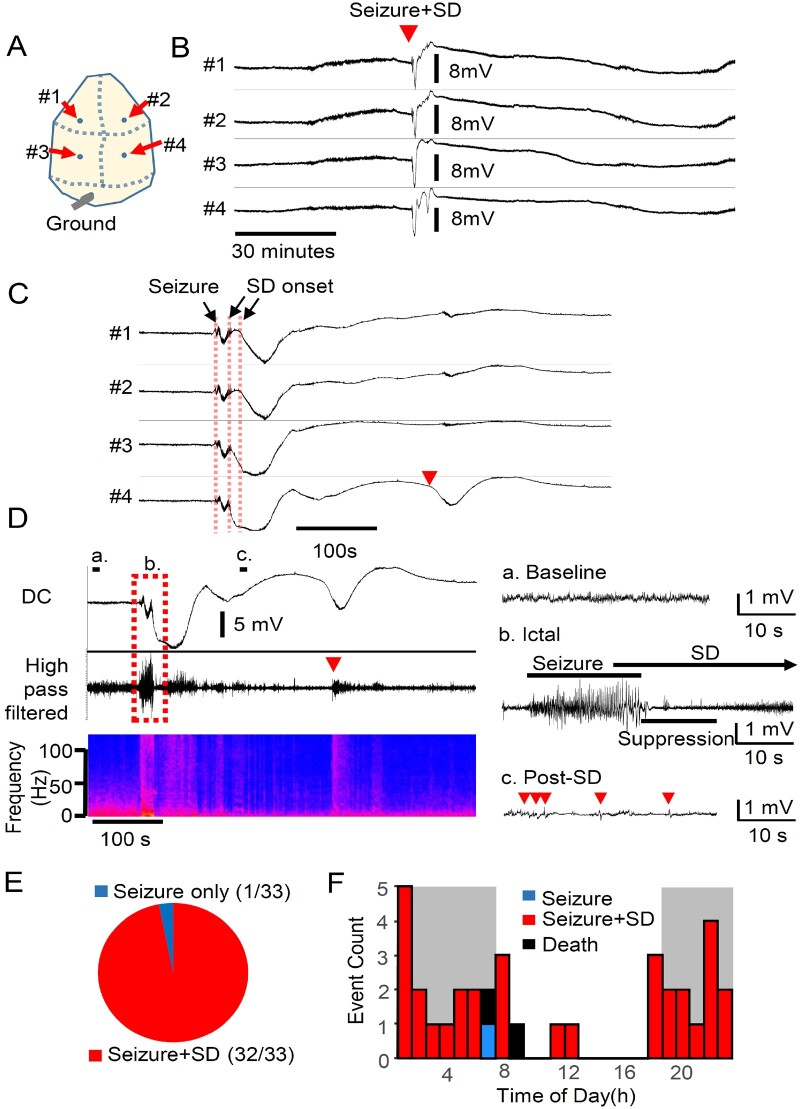
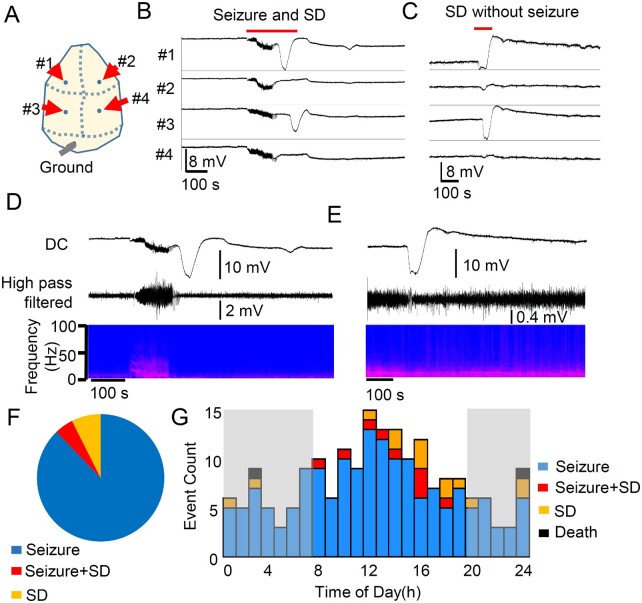
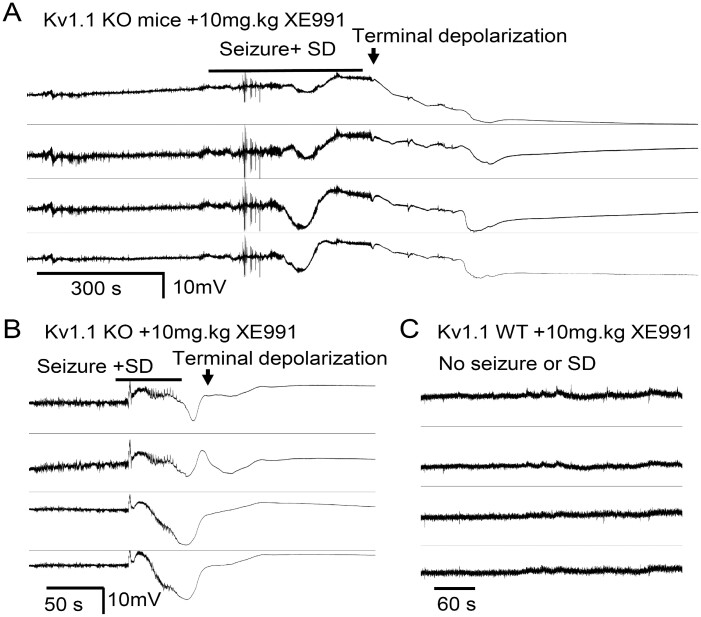
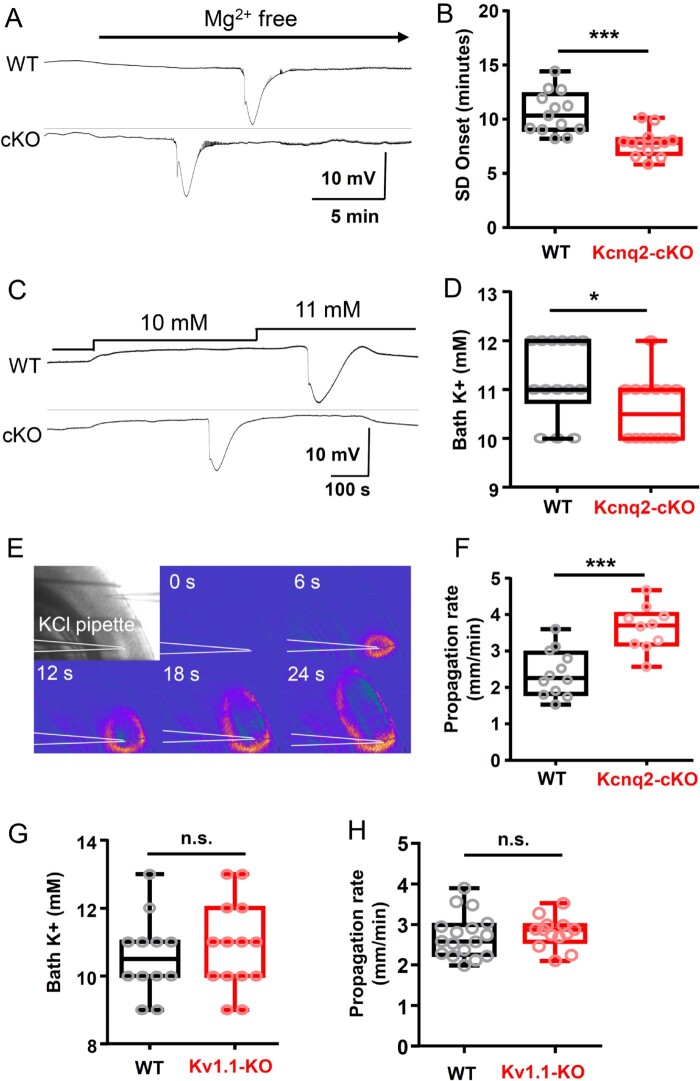
References
-
- Somjen GG. Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol Rev. 2001;81(3):1065–1096. - PubMed
-
- Dreier JP. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat Med. 2011;17(4):439–447. - PubMed
-
- Carlson AP, Shuttleworth CW, Major S, Lemale CL, Dreier JP, Hartings JA.. Terminal spreading depolarizations causing electrocortical silencing prior to clinical brain death: Case report. J Neurosurg. 2018;131(6):1773–1779. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
