

The evolutionary dynamics of endemic human coronaviruses
- PMID: 33768964
- PMCID: PMC7980080
- DOI: 10.1093/ve/veab020
The evolutionary dynamics of endemic human coronaviruses
Abstract
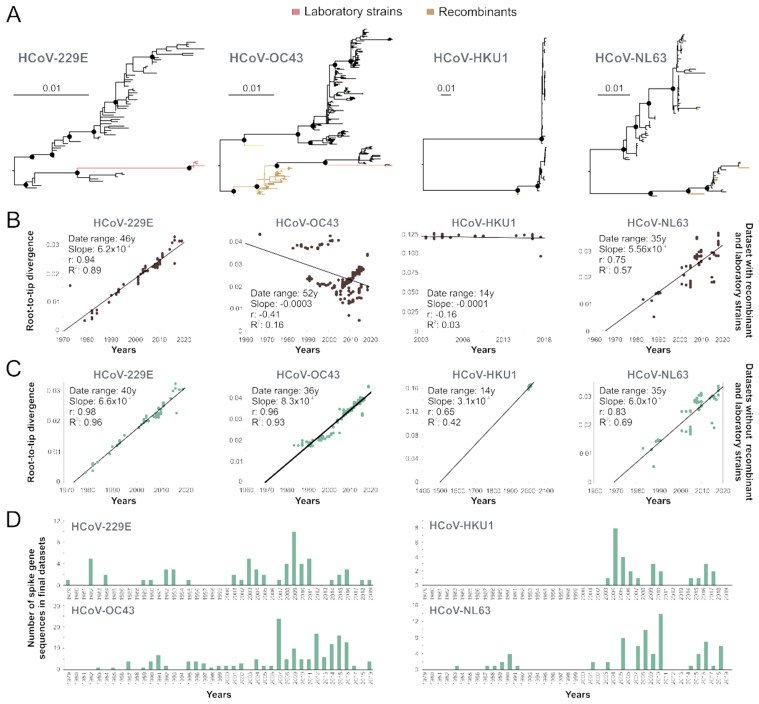
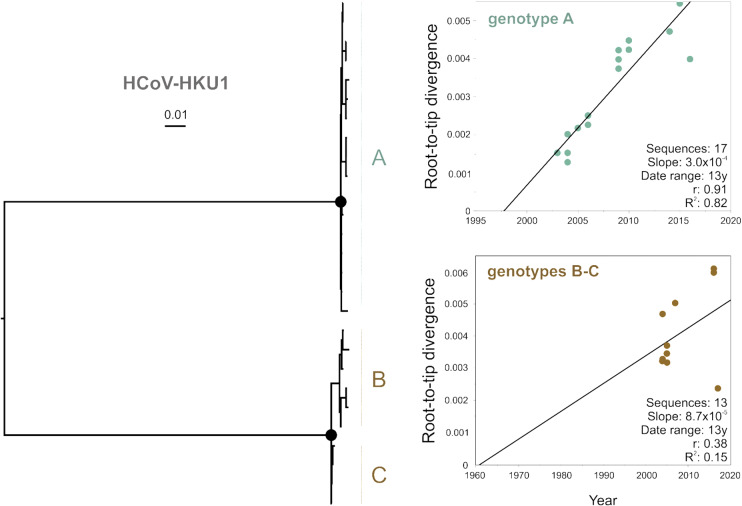
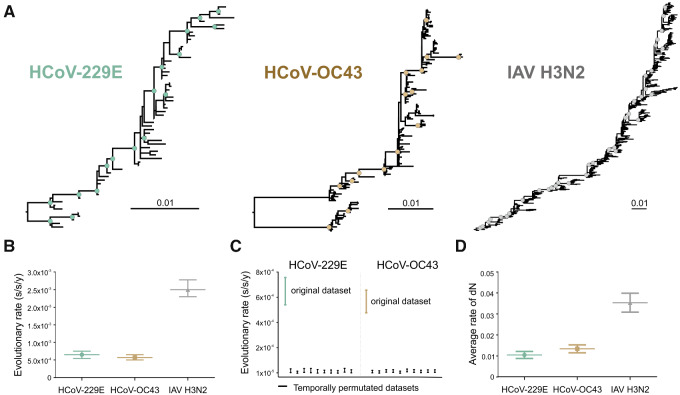
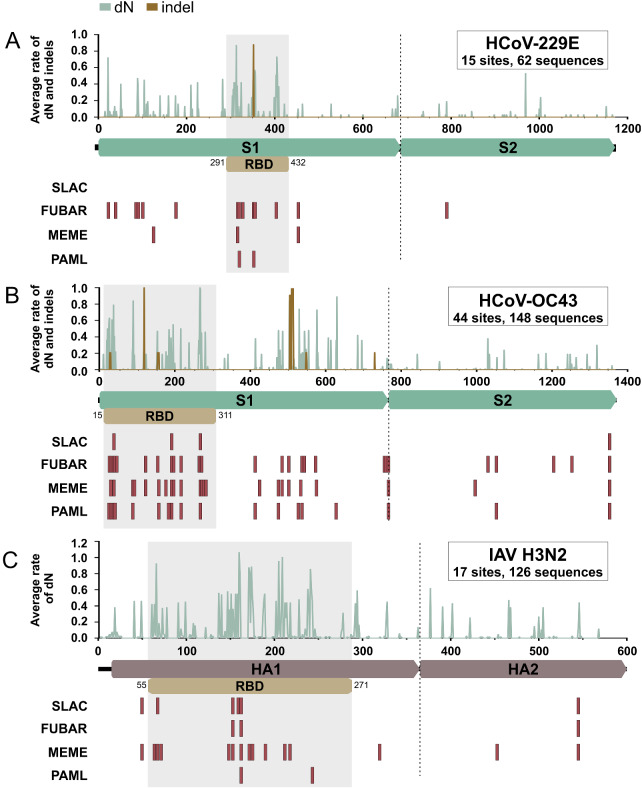
Community protective immunity can affect RNA virus evolution by selecting for new antigenic variants on the scale of years, exemplified by the need of annual evaluation of influenza vaccines. The extent to which this process termed antigenic drift affects coronaviruses remains unknown. Alike the severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), seasonal human coronaviruses (HCoV) likely emerged from animal reservoirs as new human pathogens in the past. We therefore analyzed the long-term evolutionary dynamics of the ubiquitous HCoV-229E and HCoV-OC43 in comparison with human influenza A virus (IAV) subtype H3N2. We focus on viral glycoprotein genes that mediate viral entry into cells and are major targets of host neutralizing antibody responses. Maximum likelihood and Bayesian phylogenies of publicly available gene datasets representing about three decades of HCoV and IAV evolution showed that all viruses had similar ladder-like tree shapes compatible with antigenic drift, supported by different tree shape statistics. Evolutionary rates inferred in a Bayesian framework were 6.5 × 10-4 (95% highest posterior density (HPD), 5.4-7.5 × 10-4) substitutions per site per year (s/s/y) for HCoV-229E spike (S) genes and 5.7 × 10-4 (95% HPD, 5-6.5 × 10-4) s/s/y for HCoV-OC43 S genes, which were about fourfold lower than the 2.5 × 10-3 (95% HPD, 2.3-2.7 × 10-3) s/s/y rate for IAV hemagglutinin (HA) genes. Coronavirus S genes accumulated about threefold less (P < 0.001) non-synonymous mutations (dN) over time than IAV HA genes. In both IAV and HCoV, the average rate of dN within the receptor binding domains (RBD) was about fivefold higher (P < 0.0001) than in other glycoprotein gene regions. Similarly, most sites showing evidence for positive selection occurred within the RBD (HCoV-229E, 6/14 sites, P < 0.05; HCoV-OC43, 23/38 sites, P < 0.01; IAV, 13/15 sites, P = 0.08). In sum, the evolutionary dynamics of HCoV and IAV showed several similarities, yet amino acid changes potentially representing antigenic drift occurred on a lower scale in endemic HCoV compared to IAV. It seems likely that pandemic SARS-CoV-2 evolution will bear similarities with IAV evolution including accumulation of adaptive changes in the RBD, requiring vaccines to be updated regularly, whereas higher SARS-CoV-2 evolutionary stability resembling endemic HCoV can be expected in the post-pandemic stage.
Keywords: evolutionary rate; genetic variability; human coronaviruses; mutations; vaccine.
© The Author(s) 2021. Published by Oxford University Press.
Figures
References
-
- Boni M. F. et al. (2020) ‘ Evolutionary Origins of the SARS-CoV-2 Sarbecovirus Lineage Responsible for the COVID-19 Pandemic’, Nature Microbiology, 5: 1408–17, - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
