

Sodium channelopathies of skeletal muscle and brain
- PMID: 33769100
- PMCID: PMC8989381
- DOI: 10.1152/physrev.00025.2020
Sodium channelopathies of skeletal muscle and brain
Abstract
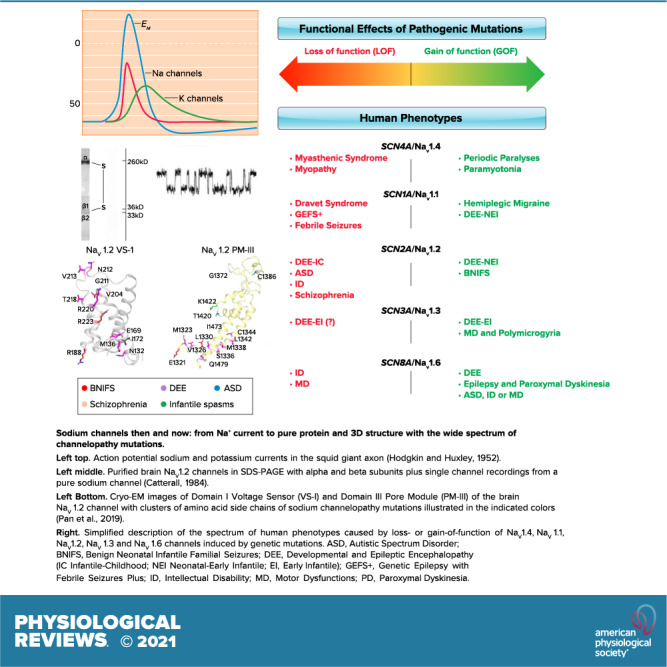
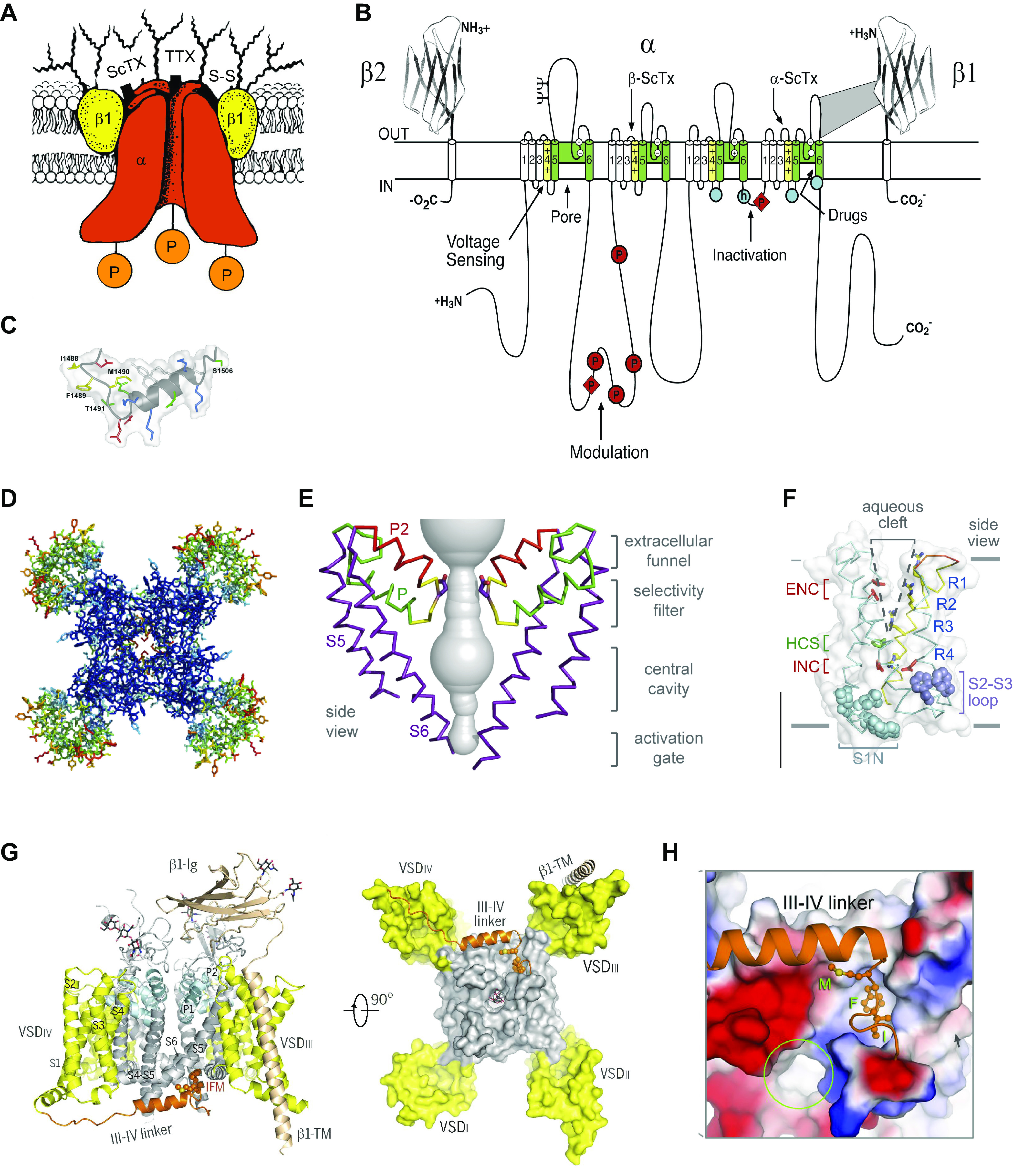
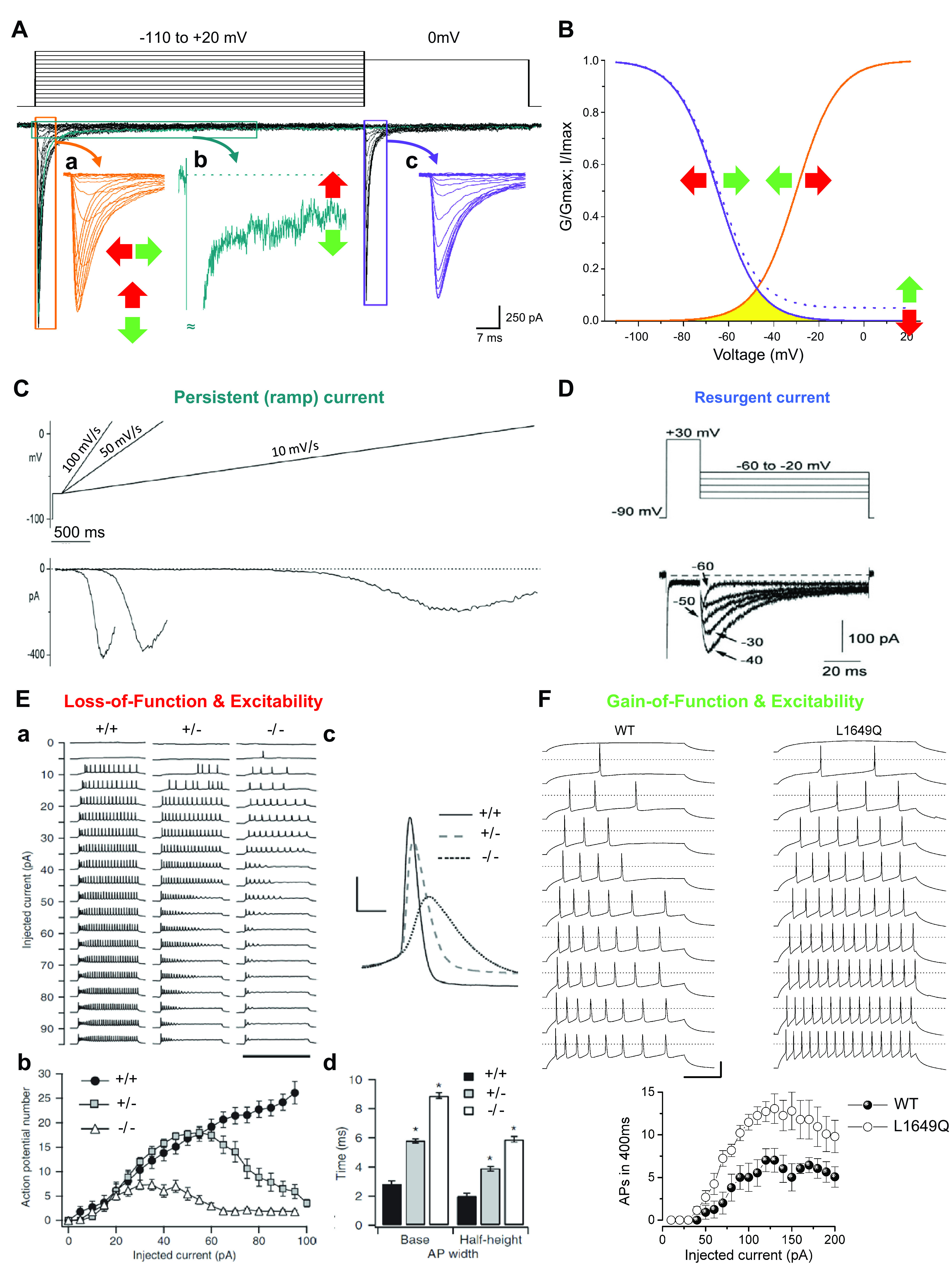
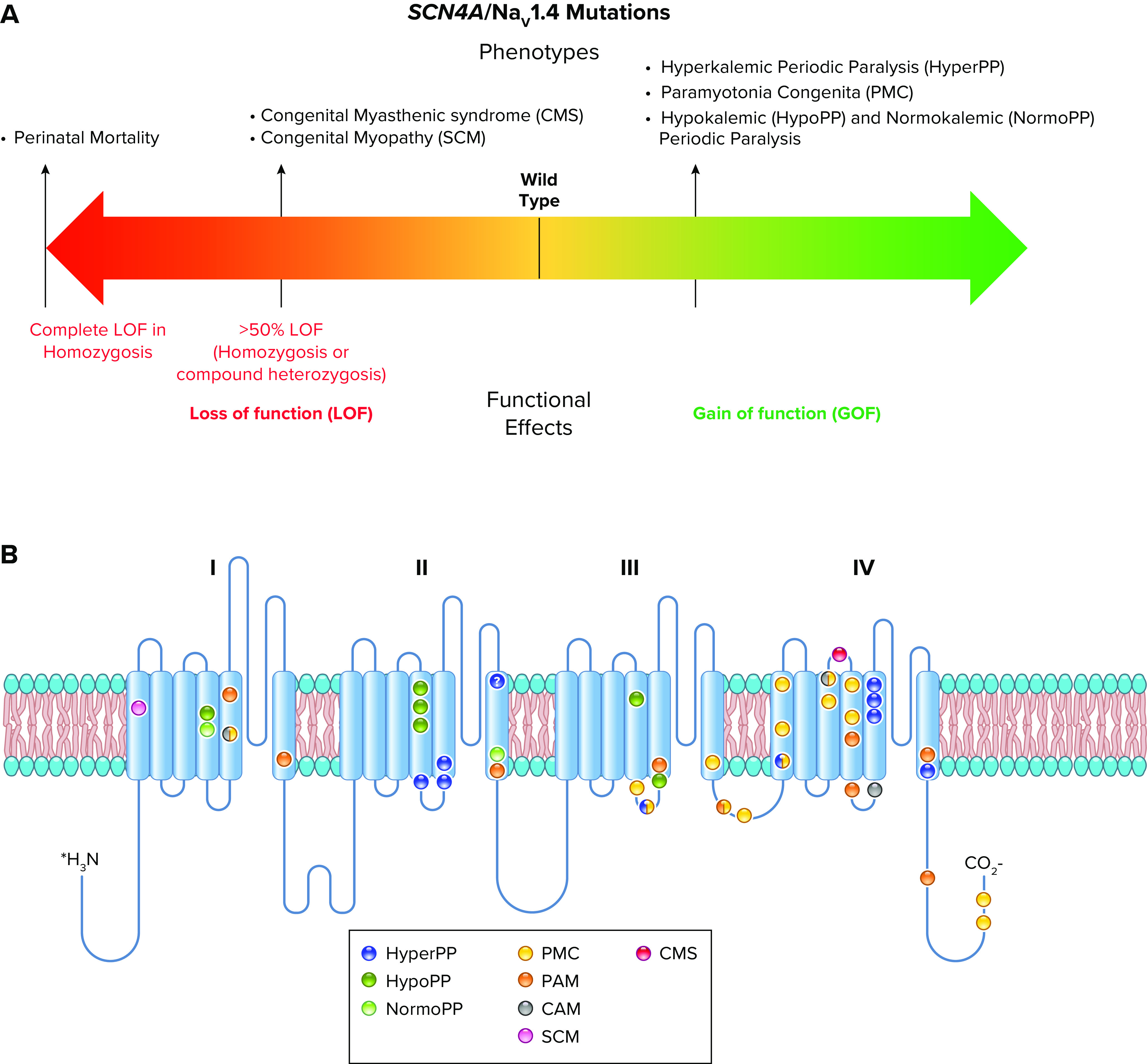
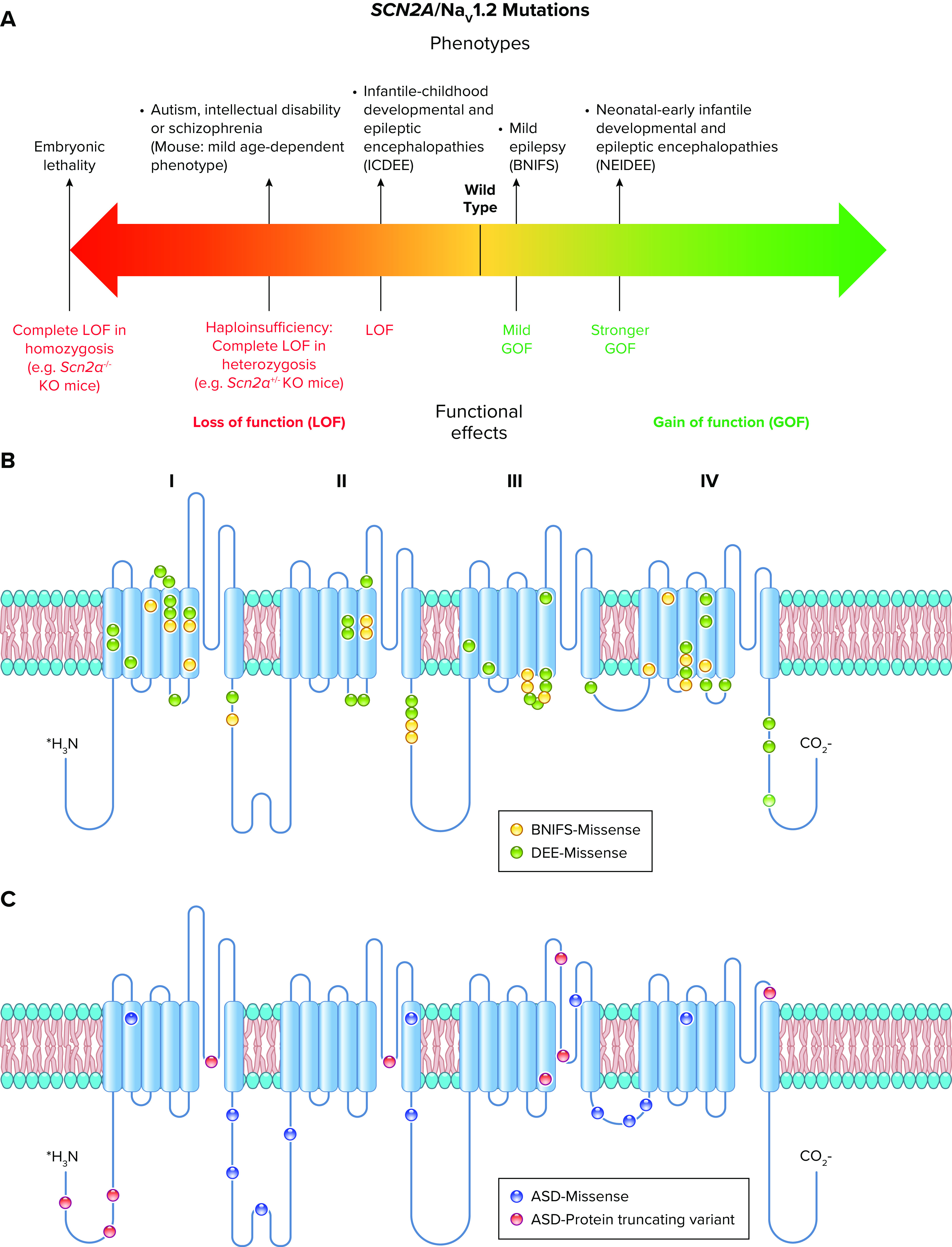
Voltage-gated sodium channels initiate action potentials in nerve, skeletal muscle, and other electrically excitable cells. Mutations in them cause a wide range of diseases. These channelopathy mutations affect every aspect of sodium channel function, including voltage sensing, voltage-dependent activation, ion conductance, fast and slow inactivation, and both biosynthesis and assembly. Mutations that cause different forms of periodic paralysis in skeletal muscle were discovered first and have provided a template for understanding structure, function, and pathophysiology at the molecular level. More recent work has revealed multiple sodium channelopathies in the brain. Here we review the well-characterized genetics and pathophysiology of the periodic paralyses of skeletal muscle and then use this information as a foundation for advancing our understanding of mutations in the structurally homologous α-subunits of brain sodium channels that cause epilepsy, migraine, autism, and related comorbidities. We include studies based on molecular and structural biology, cell biology and physiology, pharmacology, and mouse genetics. Our review reveals unexpected connections among these different types of sodium channelopathies.
Keywords: autism; epilepsy; migraine; periodic paralysis; sodium channels.
Conflict of interest statement
No conflicts of interest, financial or otherwise, are declared by the authors.
Figures
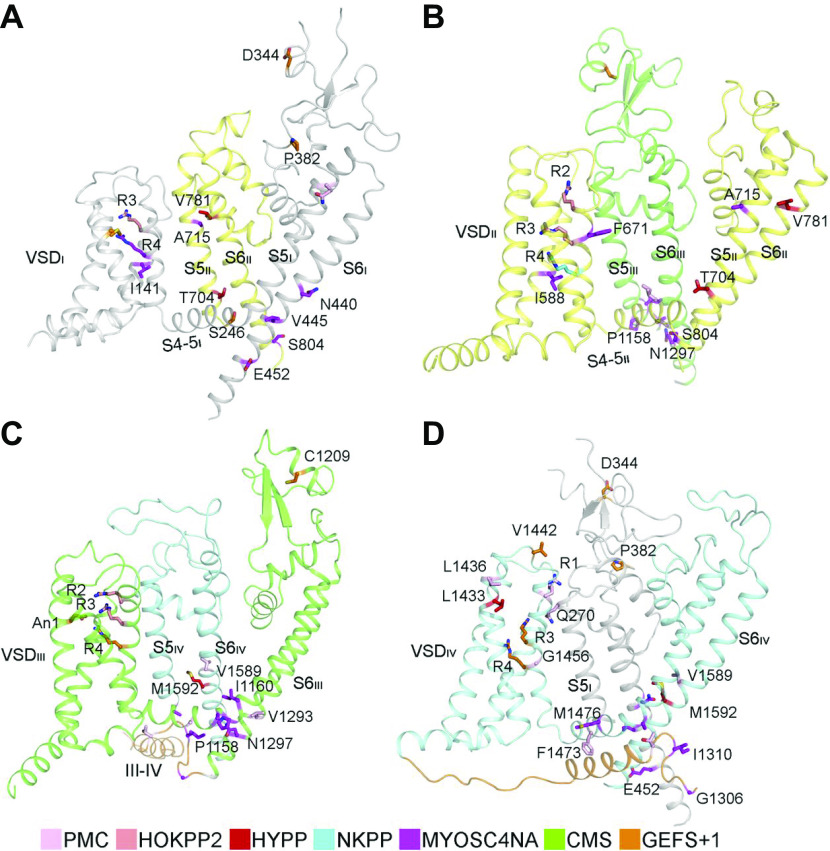
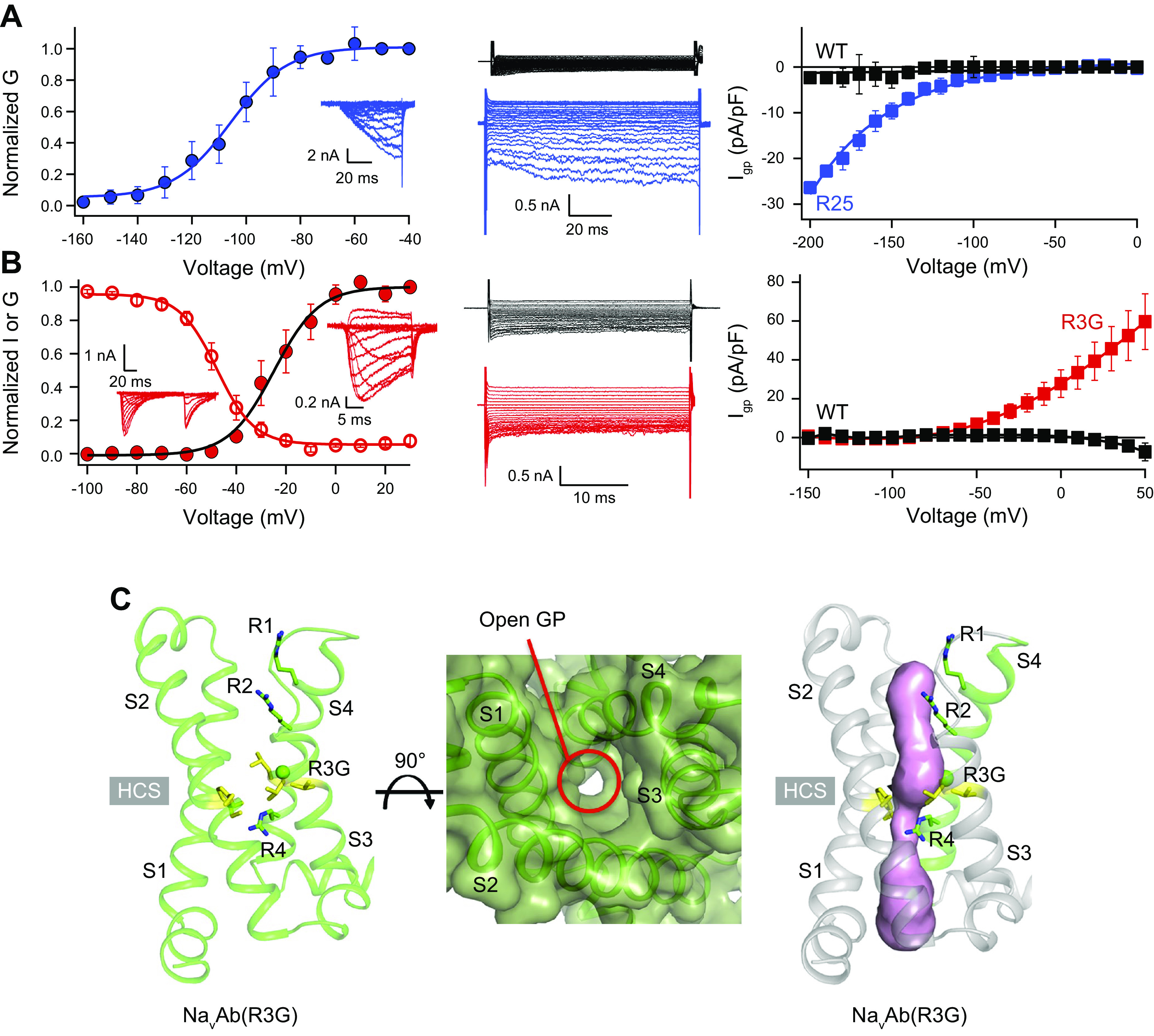
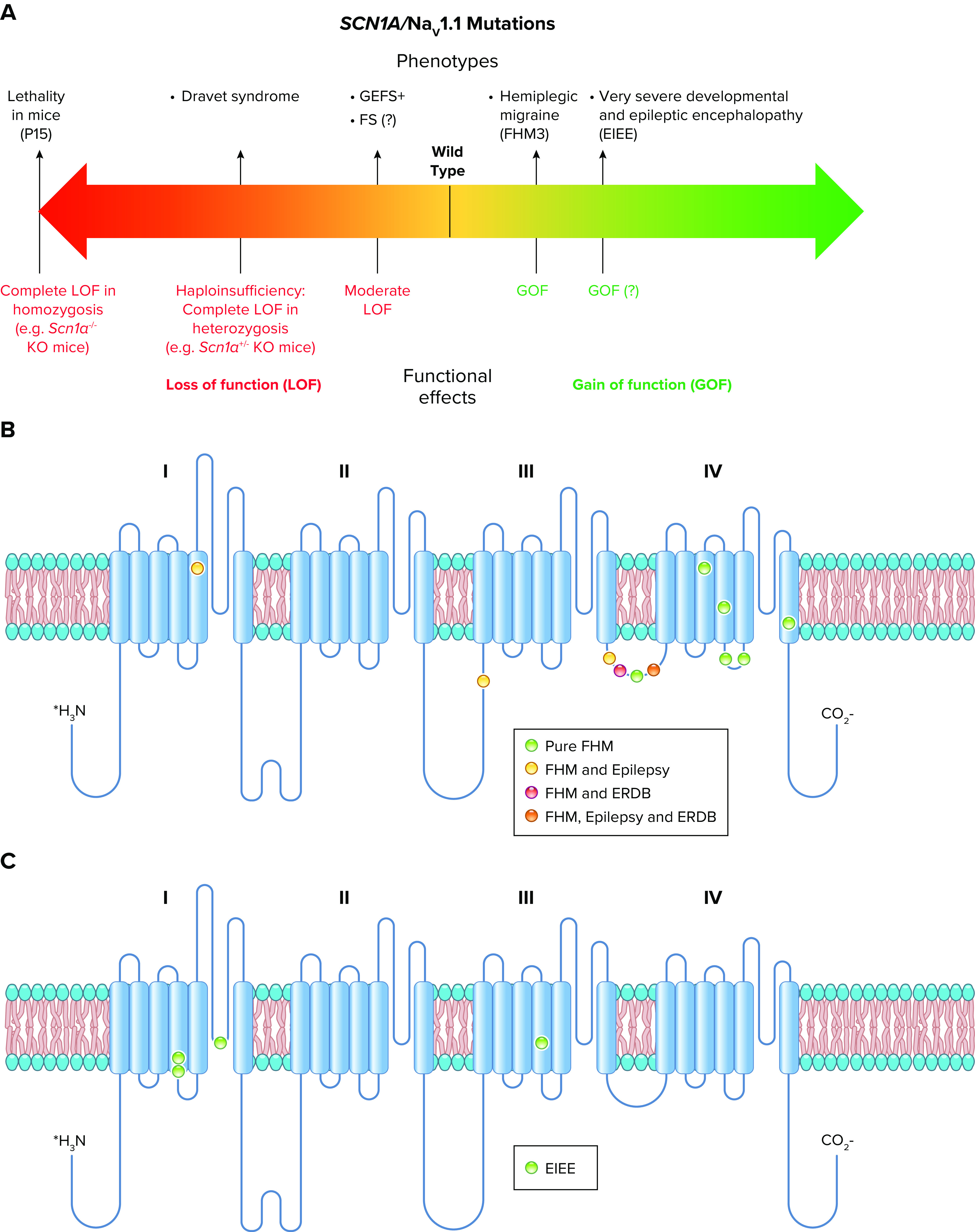
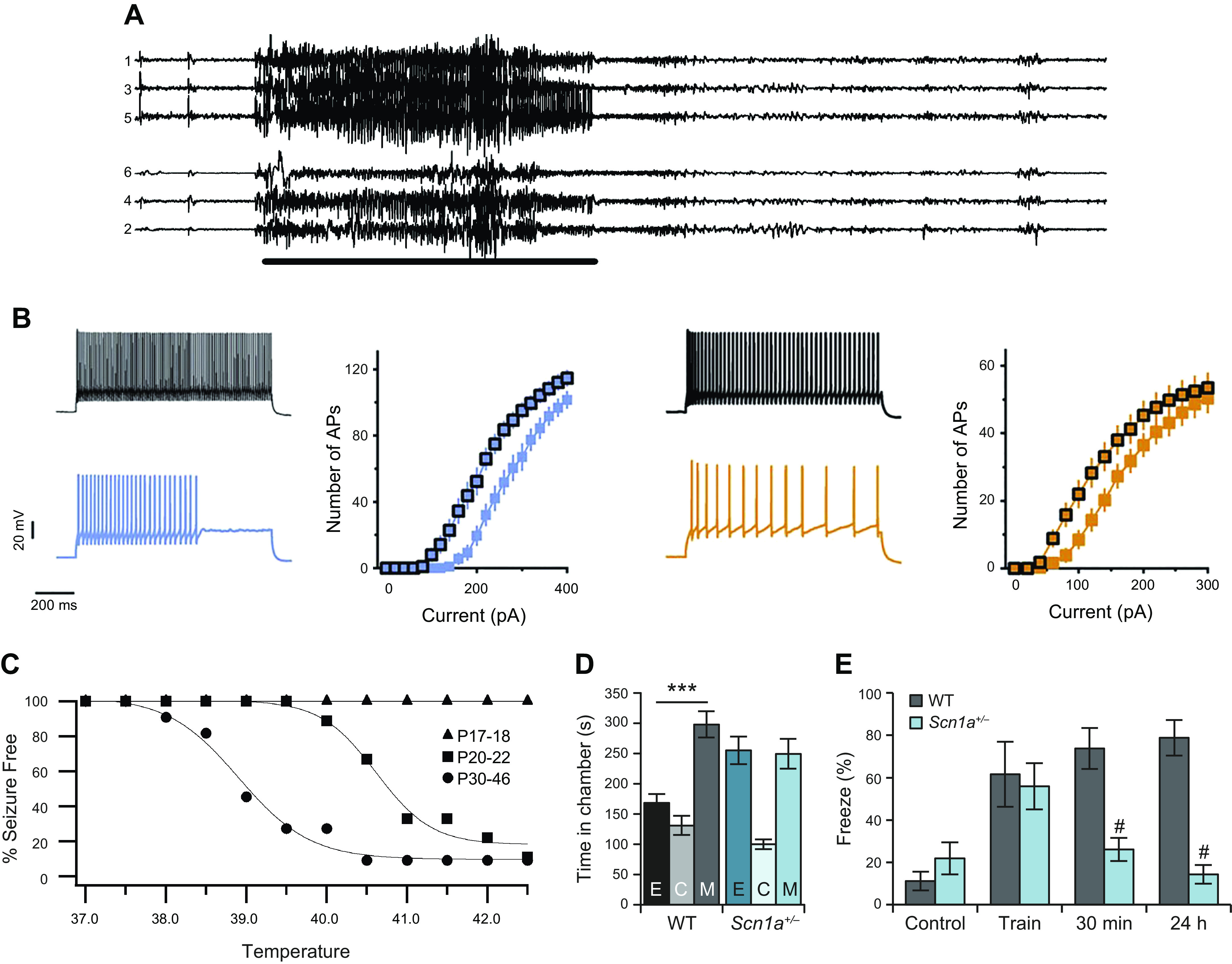
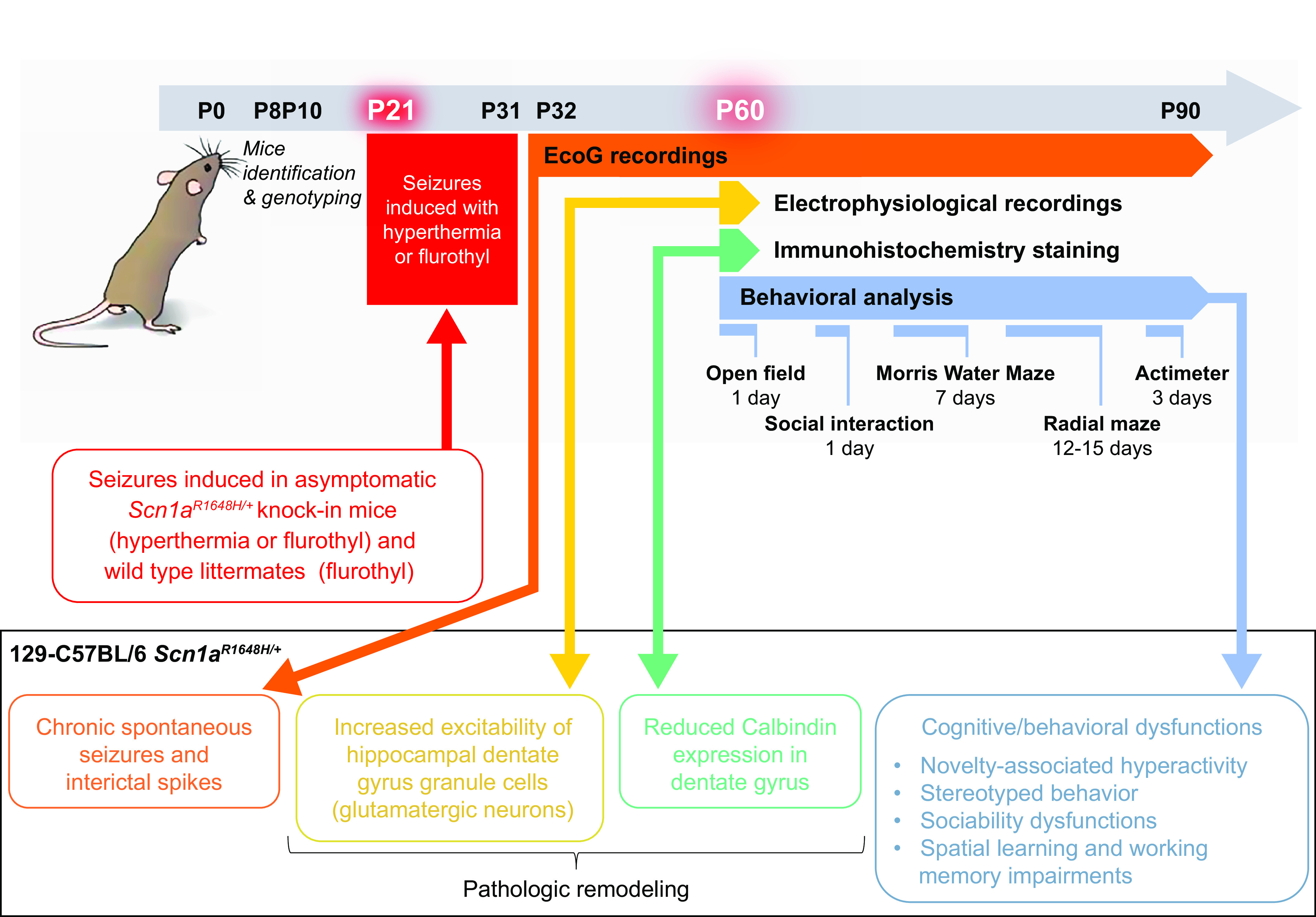
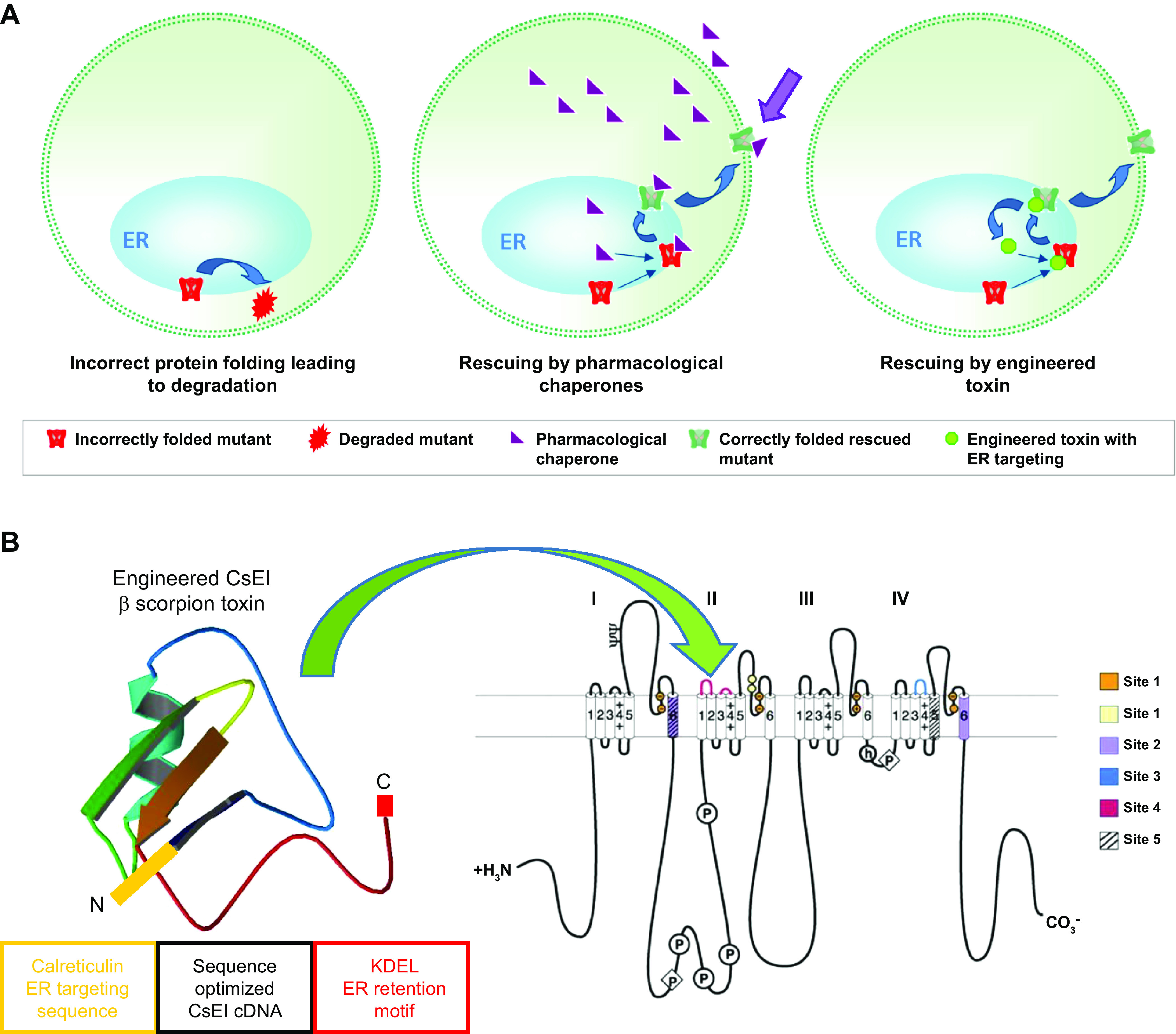
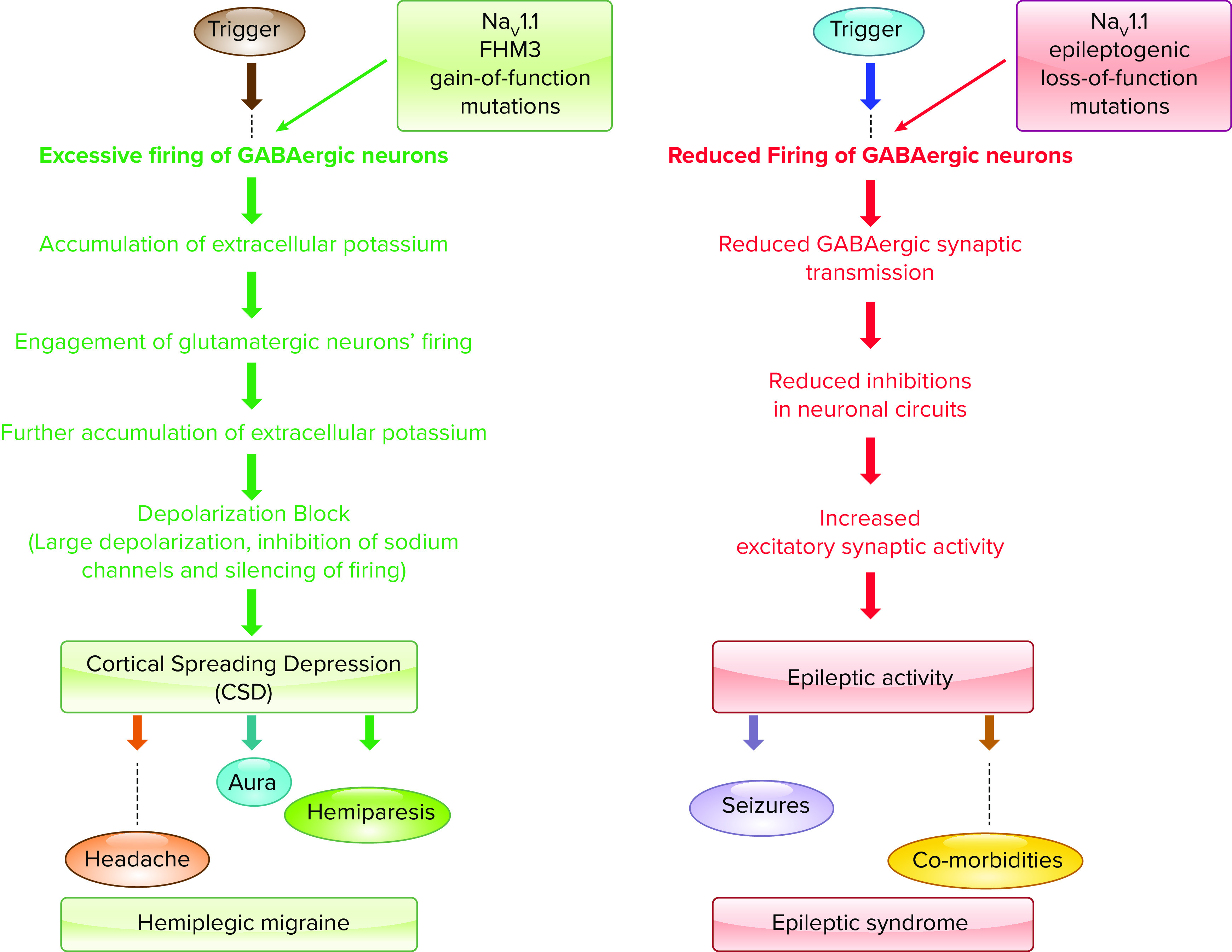
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources