

Microbial genetic and transcriptional contributions to oxalate degradation by the gut microbiota in health and disease
- PMID: 33769280
- PMCID: PMC8062136
- DOI: 10.7554/eLife.63642
Microbial genetic and transcriptional contributions to oxalate degradation by the gut microbiota in health and disease
Abstract
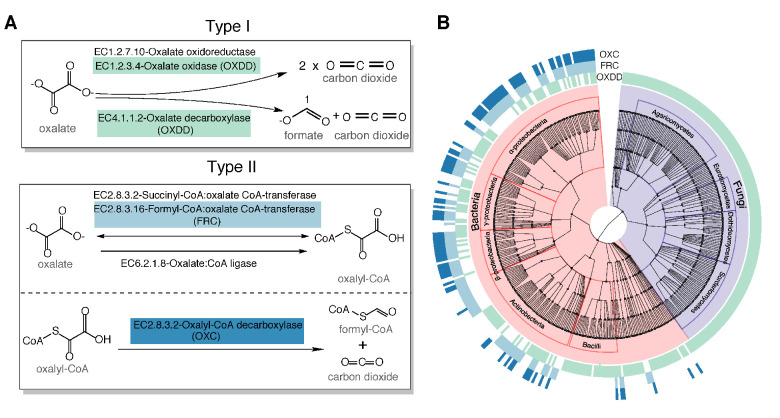
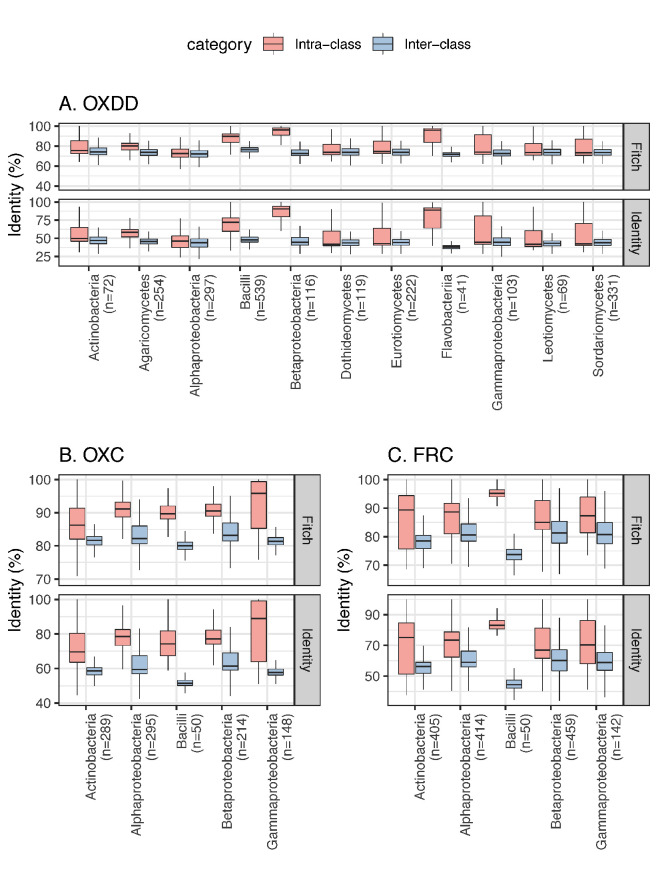
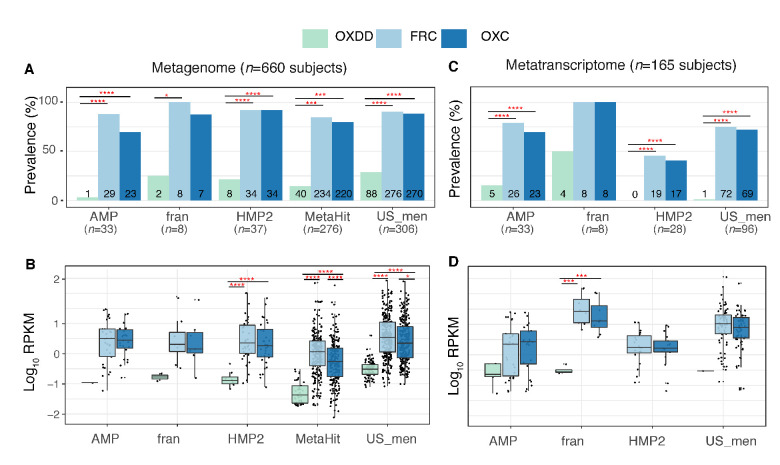
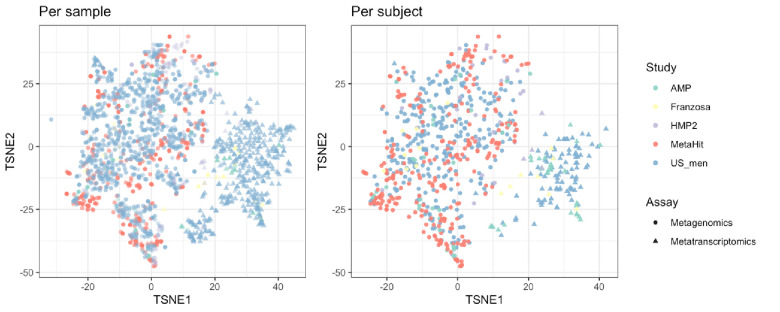
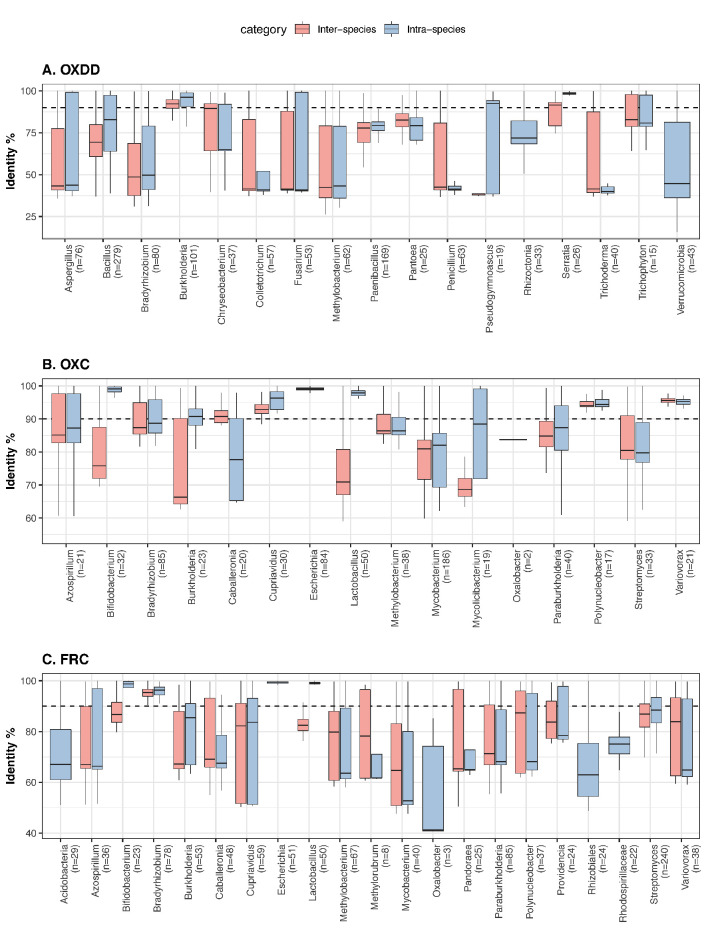
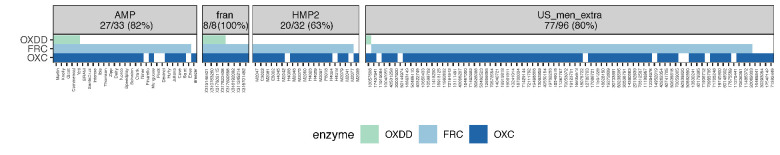
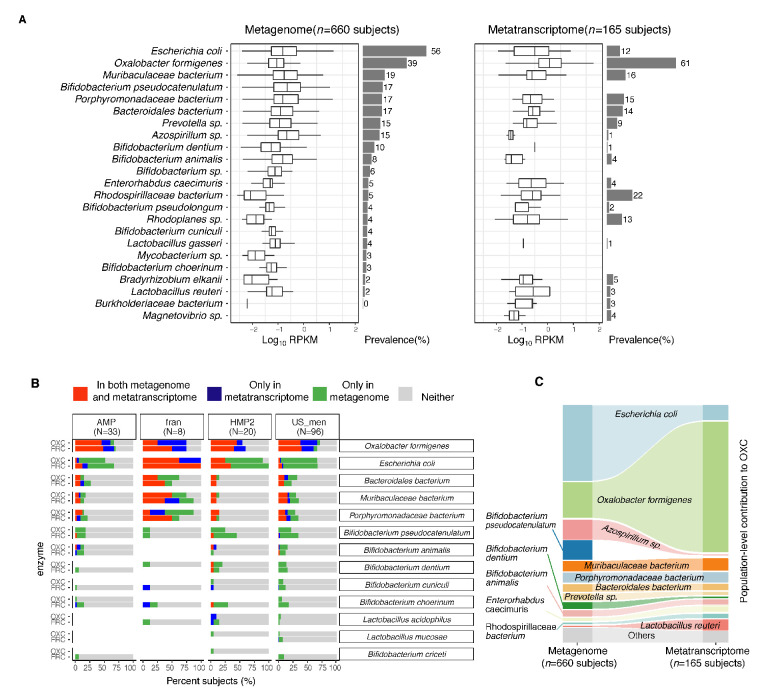
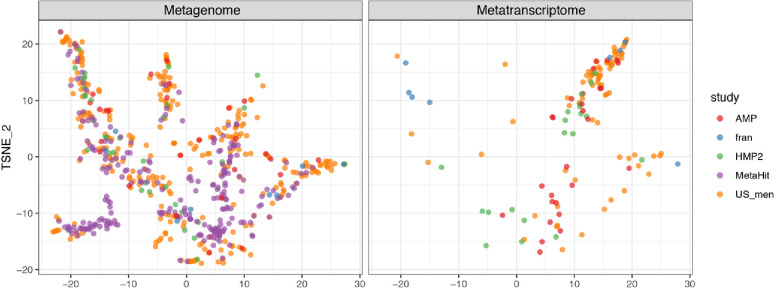
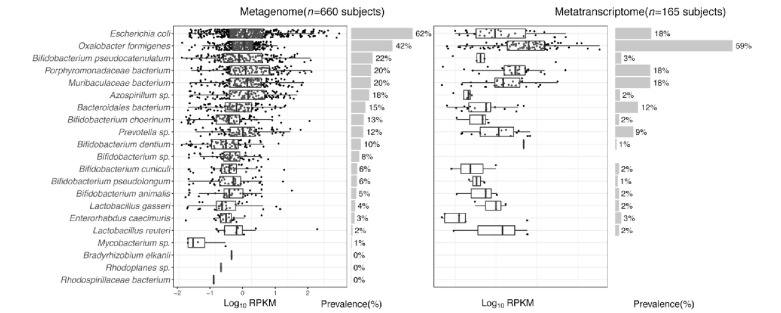
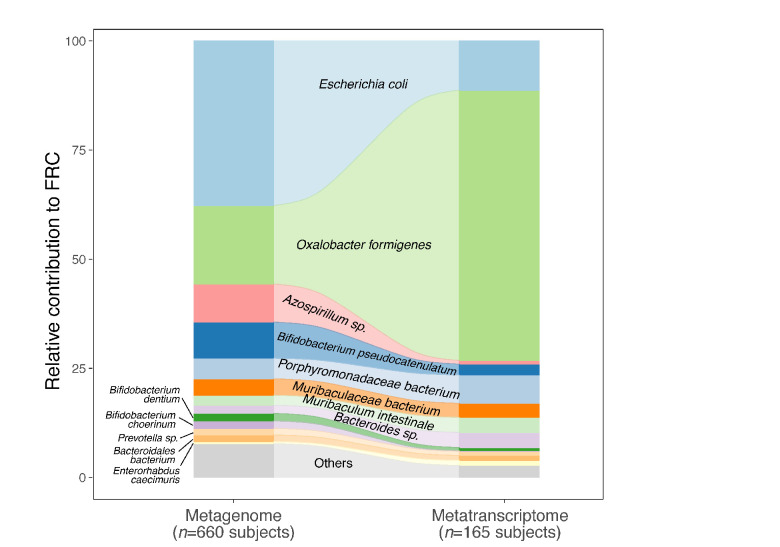
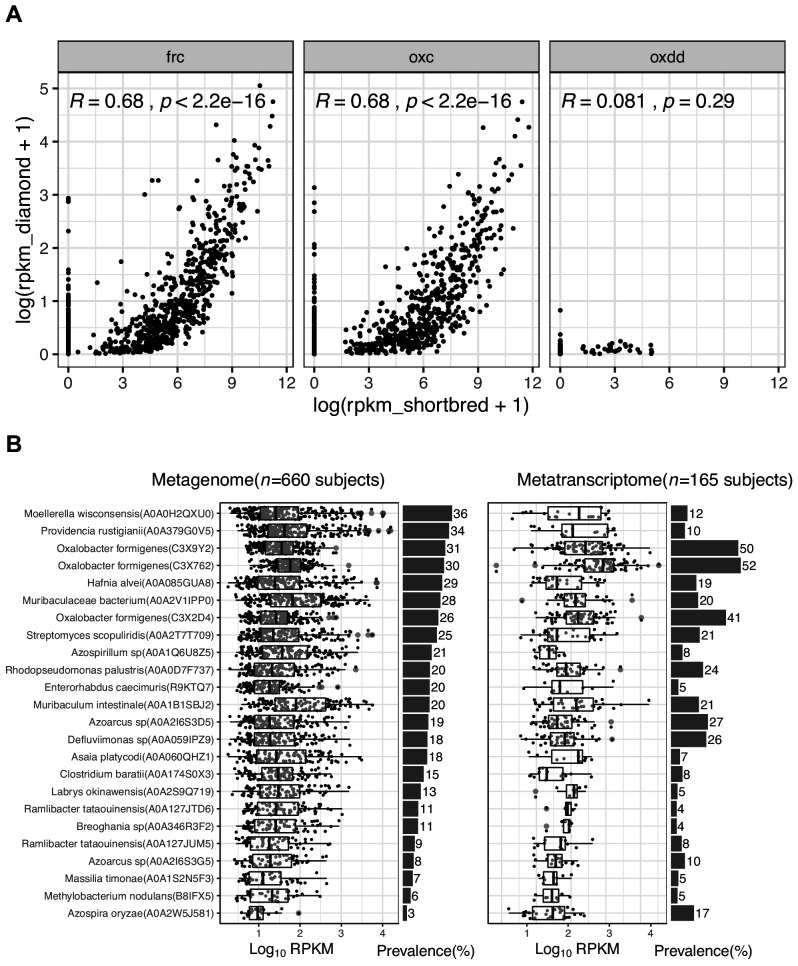
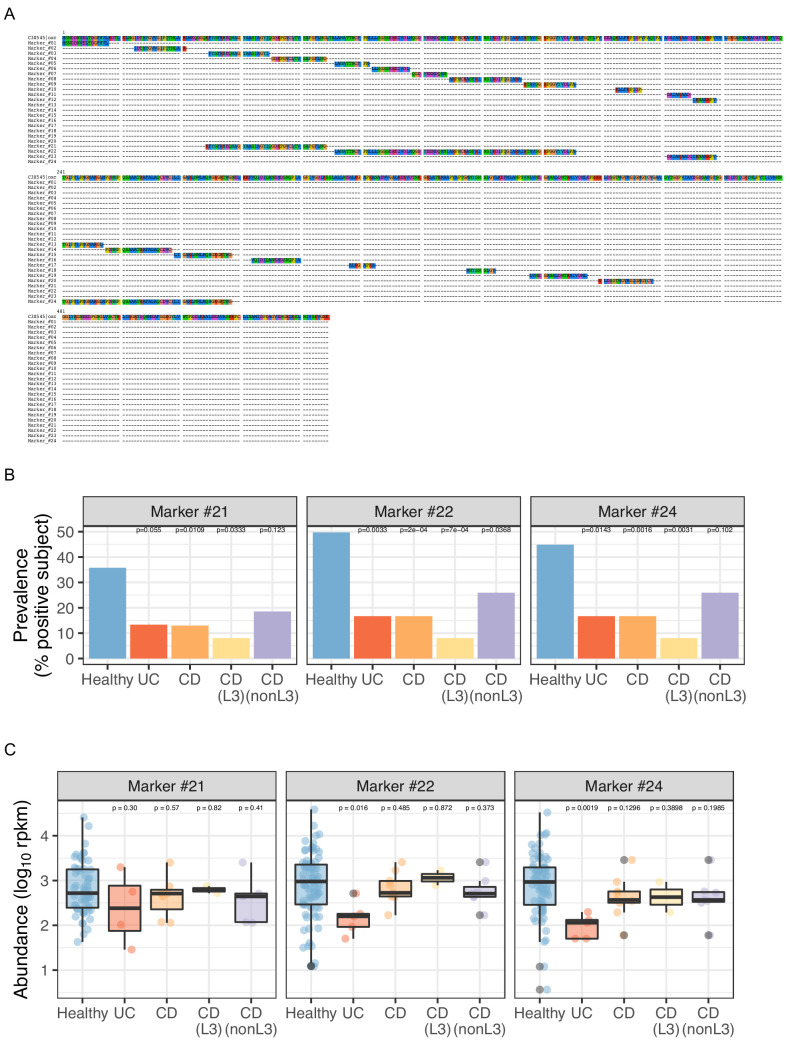
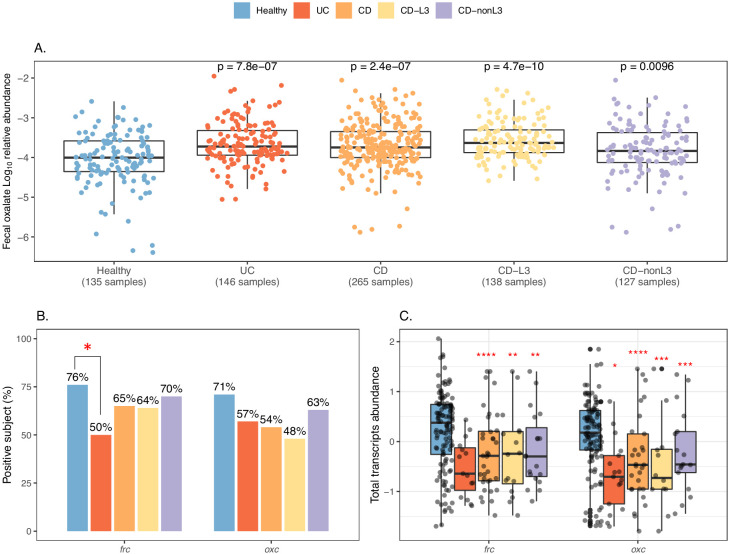
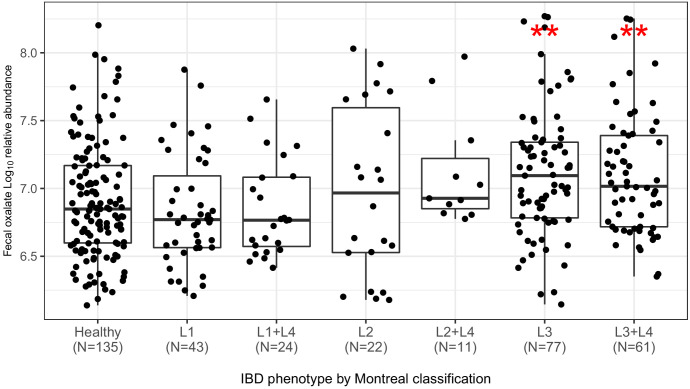
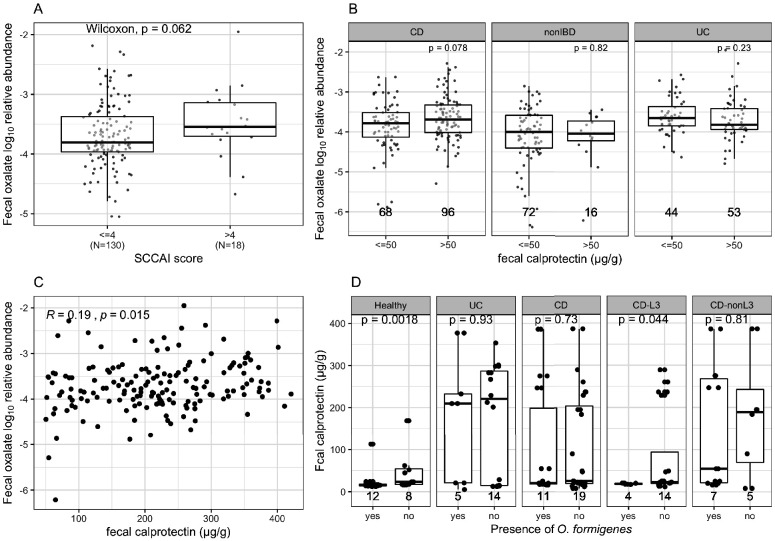
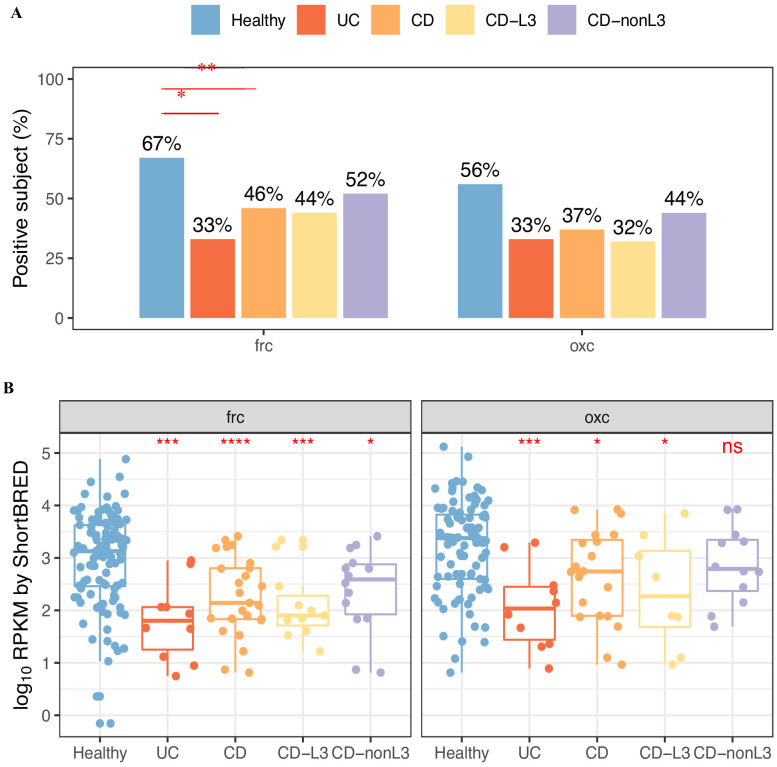
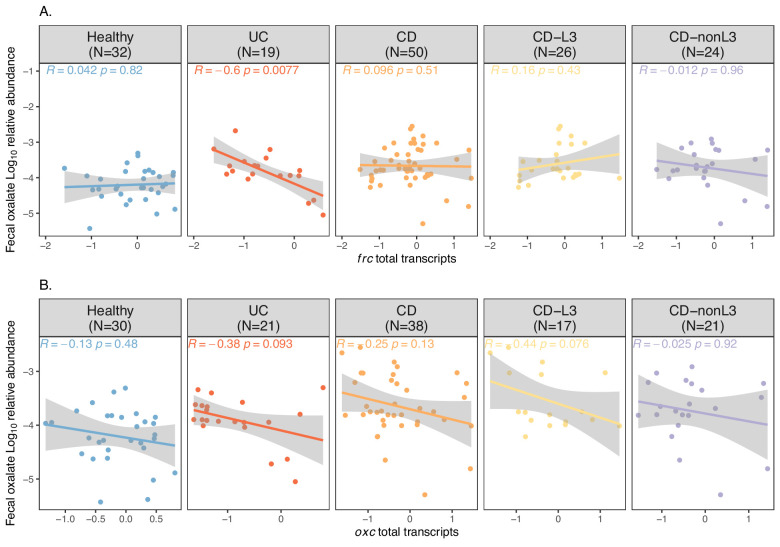
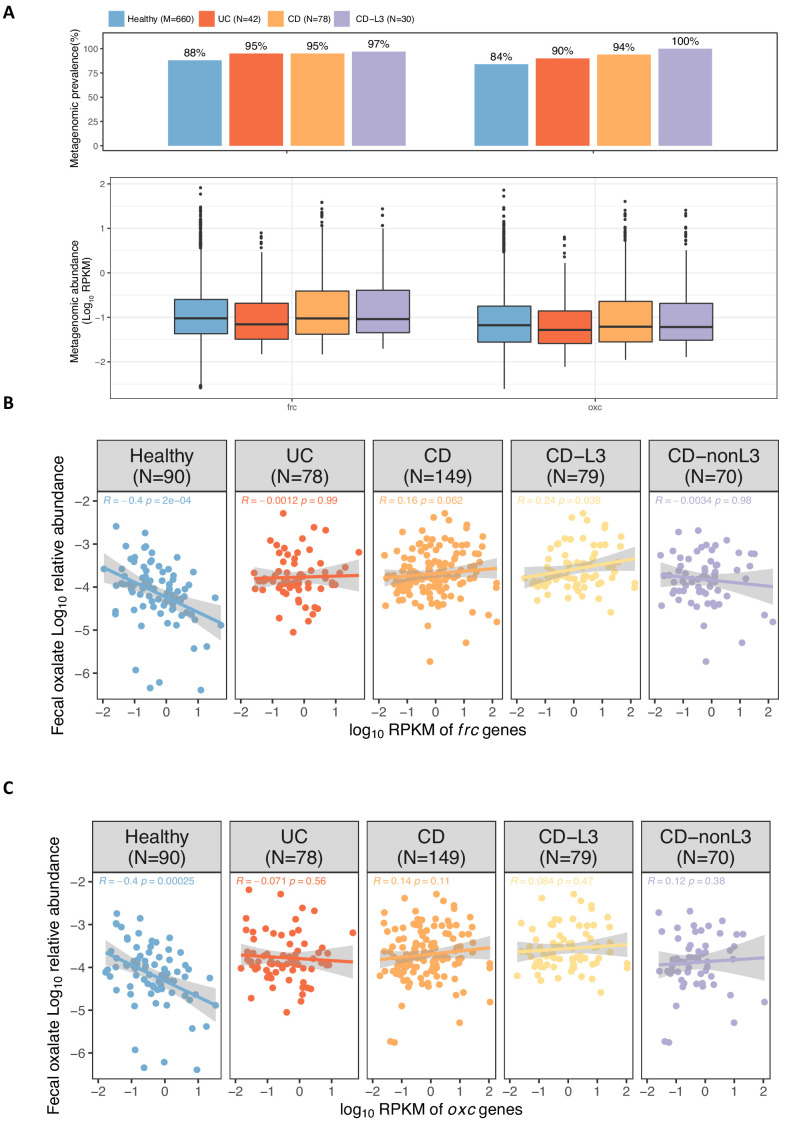
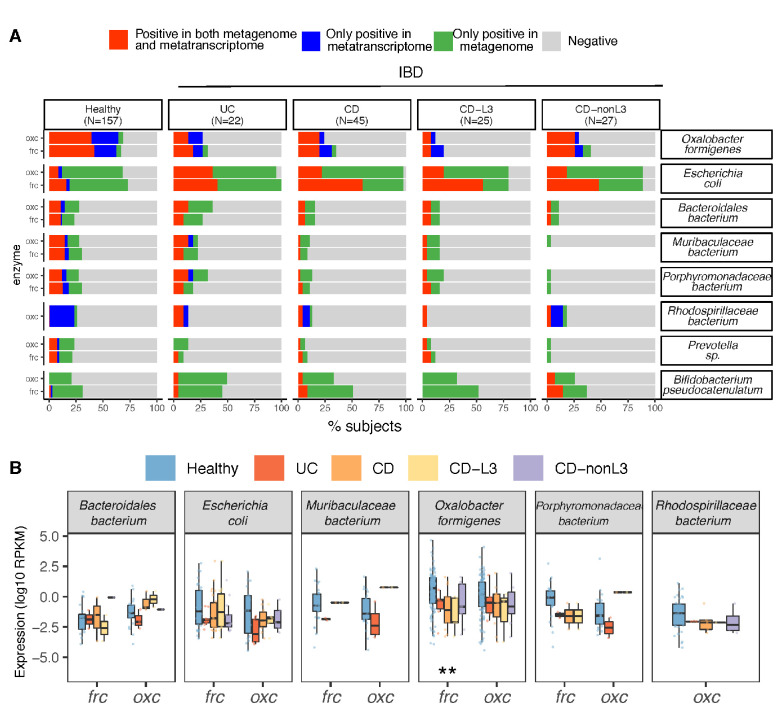
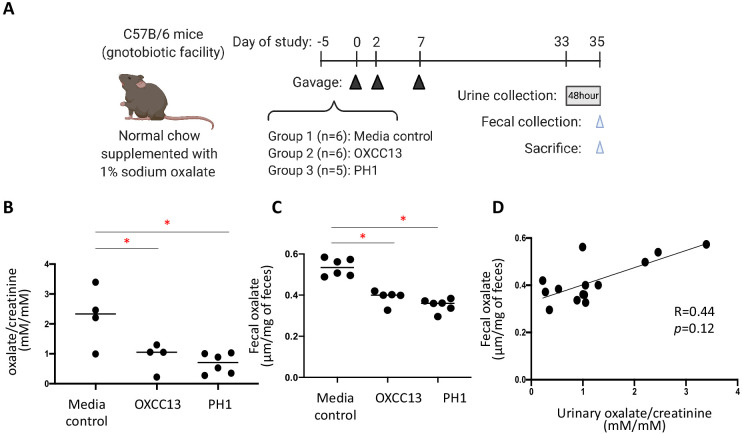
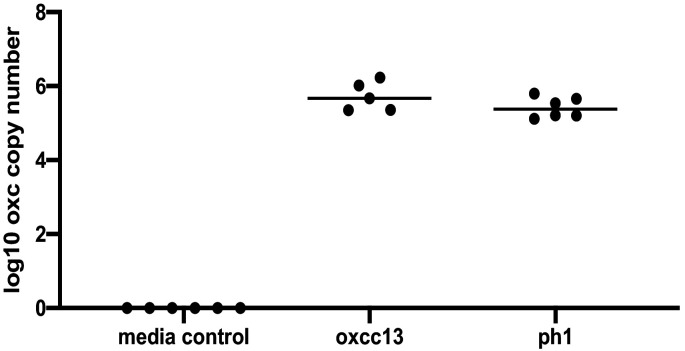
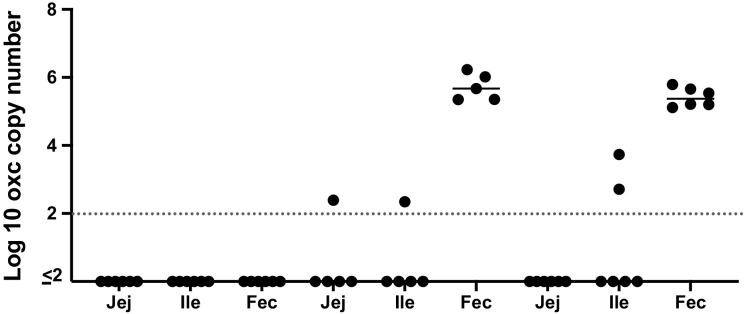
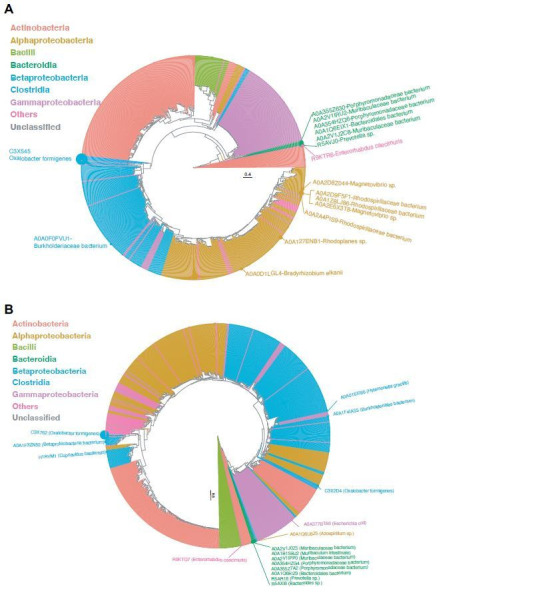
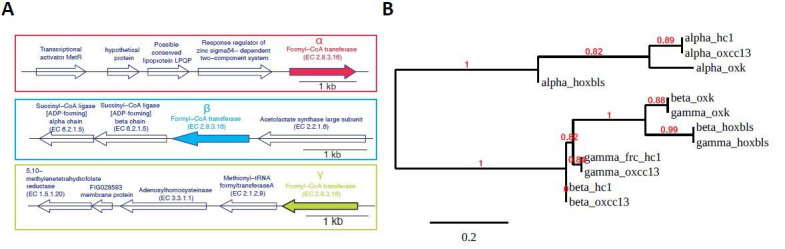
Over-accumulation of oxalate in humans may lead to nephrolithiasis and nephrocalcinosis. Humans lack endogenous oxalate degradation pathways (ODP), but intestinal microbes can degrade oxalate using multiple ODPs and protect against its absorption. The exact oxalate-degrading taxa in the human microbiota and their ODP have not been described. We leverage multi-omics data (>3000 samples from >1000 subjects) to show that the human microbiota primarily uses the type II ODP, rather than type I. Furthermore, among the diverse ODP-encoding microbes, an oxalate autotroph, Oxalobacter formigenes, dominates this function transcriptionally. Patients with inflammatory bowel disease (IBD) frequently suffer from disrupted oxalate homeostasis and calcium oxalate nephrolithiasis. We show that the enteric oxalate level is elevated in IBD patients, with highest levels in Crohn's disease (CD) patients with both ileal and colonic involvement consistent with known nephrolithiasis risk. We show that the microbiota ODP expression is reduced in IBD patients, which may contribute to the disrupted oxalate homeostasis. The specific changes in ODP expression by several important taxa suggest that they play distinct roles in IBD-induced nephrolithiasis risk. Lastly, we colonize mice that are maintained in the gnotobiotic facility with O. formigenes, using either a laboratory isolate or an isolate we cultured from human stools, and observed a significant reduction in host fecal and urine oxalate levels, supporting our in silico prediction of the importance of the microbiome, particularly O. formigenes in host oxalate homeostasis.
Keywords: IBD; computational biology; gene expressions; human; infectious disease; metagenome; metatranscriptome; microbiology; microbiota; mouse; oxalate metabolism; systems biology.
© 2021, Liu et al.
Conflict of interest statement
ML, JD, JH, AV, TB, MH, PL, HL, KR, AT, MB, LN No competing interests declared, JA is an employee of Litholink, AB is an employee of Genentech
Figures

References
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
