

Implicit Solvents for the Polarizable Atomic Multipole AMOEBA Force Field
- PMID: 33769814
- PMCID: PMC8126468
- DOI: 10.1021/acs.jctc.0c01286
Implicit Solvents for the Polarizable Atomic Multipole AMOEBA Force Field
Abstract
Computational protein design, ab initio protein/RNA folding, and protein-ligand screening can be too computationally demanding for explicit treatment of solvent. For these applications, implicit solvent offers a compelling alternative, which we describe here for the polarizable atomic multipole AMOEBA force field based on three treatments of continuum electrostatics: numerical solutions to the nonlinear and linearized versions of the Poisson-Boltzmann equation (PBE), the domain-decomposition conductor-like screening model (ddCOSMO) approximation to the PBE, and the analytic generalized Kirkwood (GK) approximation. The continuum electrostatics models are combined with a nonpolar estimator based on novel cavitation and dispersion terms. Electrostatic model parameters are numerically optimized using a least-squares style target function based on a library of 103 small-molecule solvation free energy differences. Mean signed errors for the adaptive Poisson-Boltzmann solver (APBS), ddCOSMO, and GK models are 0.05, 0.00, and 0.00 kcal/mol, respectively, while the mean unsigned errors are 0.70, 0.63, and 0.58 kcal/mol, respectively. Validation of the electrostatic response of the resulting implicit solvents, which are available in the Tinker (or Tinker-HP), OpenMM, and Force Field X software packages, is based on comparisons to explicit solvent simulations for a series of proteins and nucleic acids. Overall, the emergence of performative implicit solvent models for polarizable force fields opens the door to their use for folding and design applications.
Figures






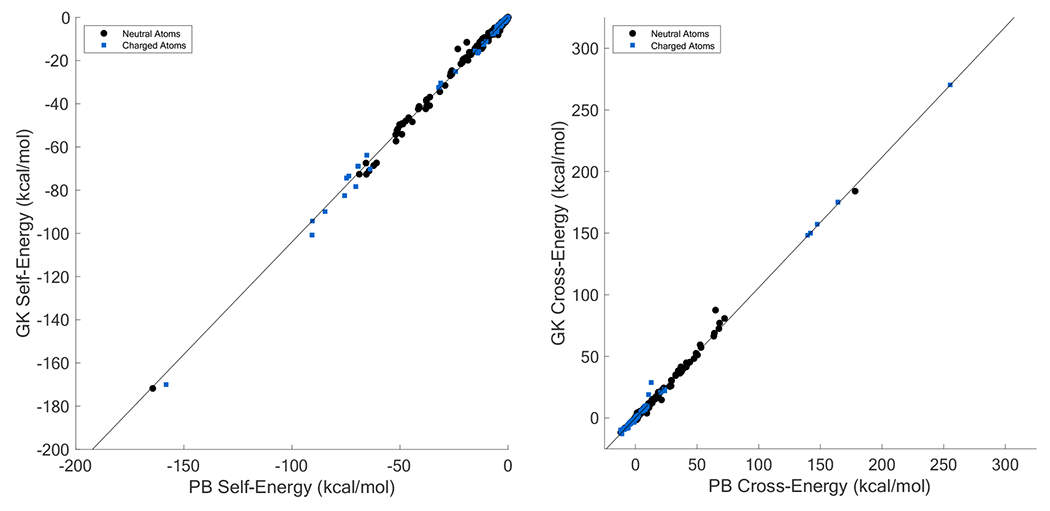


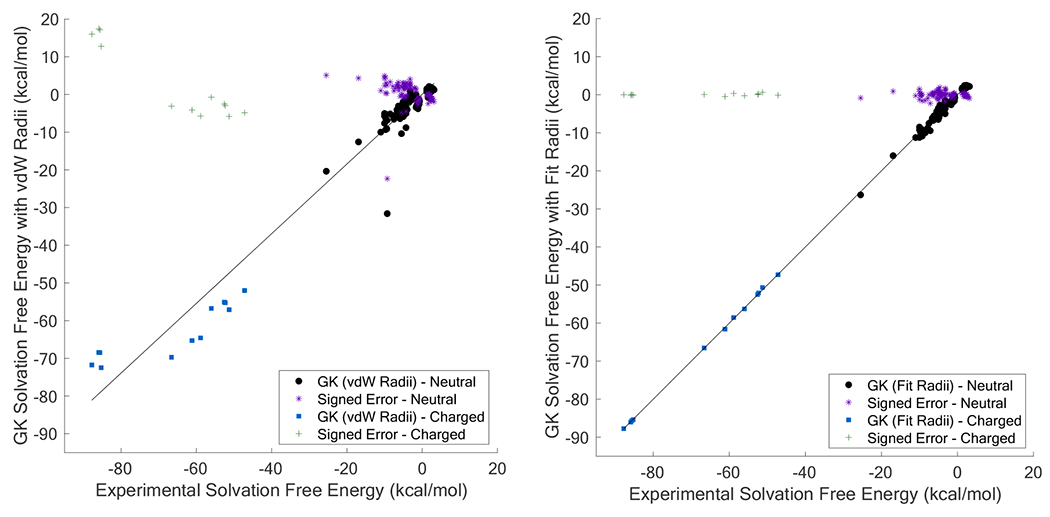




References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous