

Regenerative cross talk between cardiac cells and macrophages
- PMID: 33769920
- PMCID: PMC8289360
- DOI: 10.1152/ajpheart.00056.2021
Regenerative cross talk between cardiac cells and macrophages
Abstract
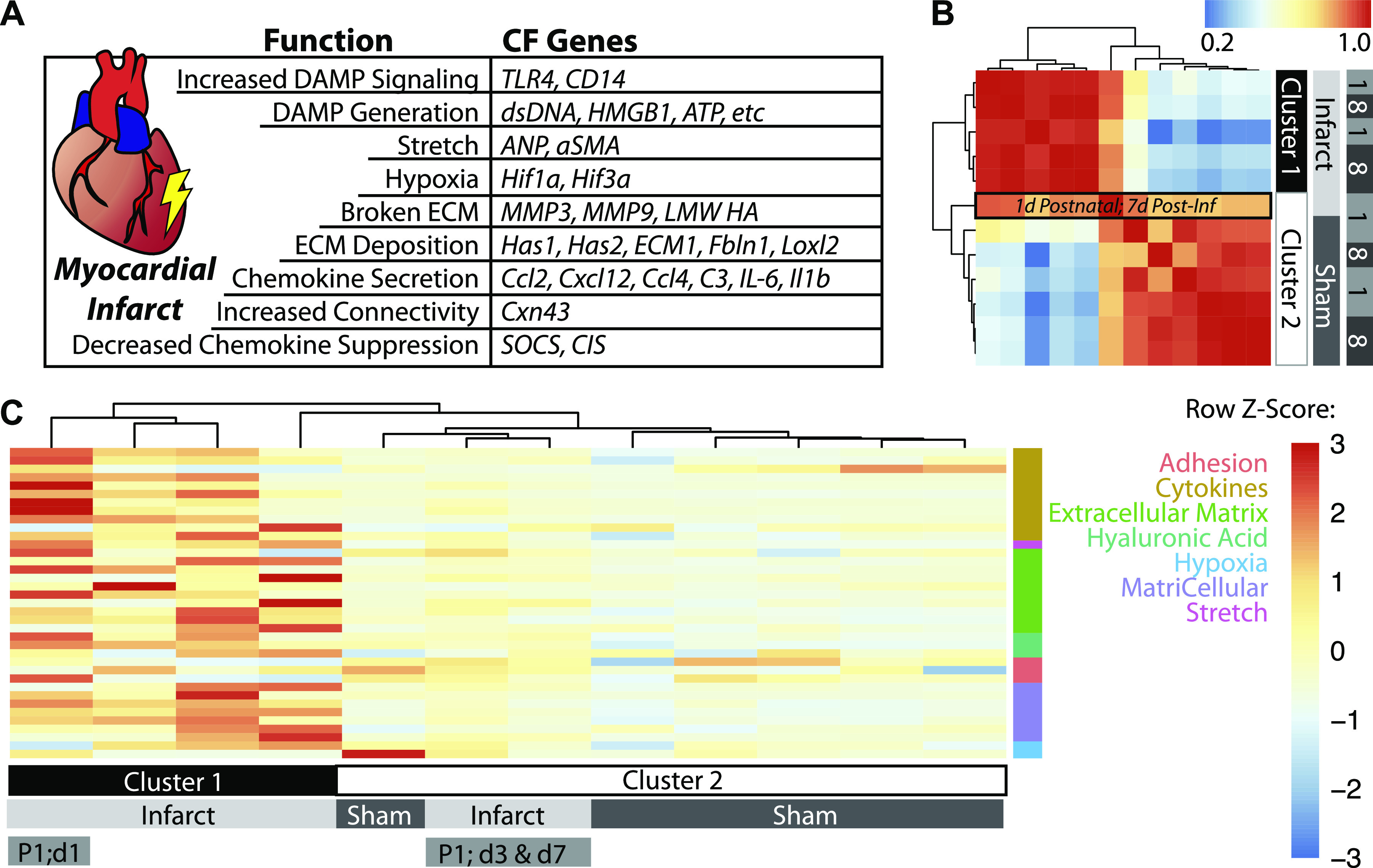
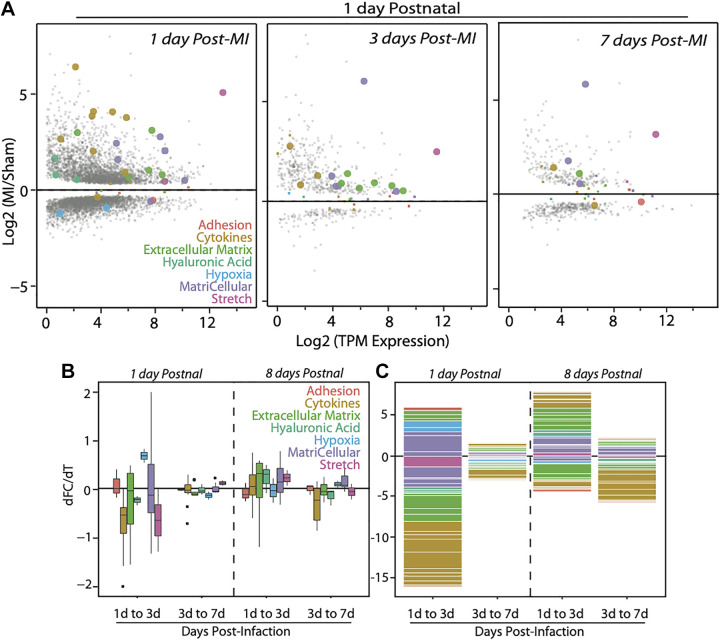
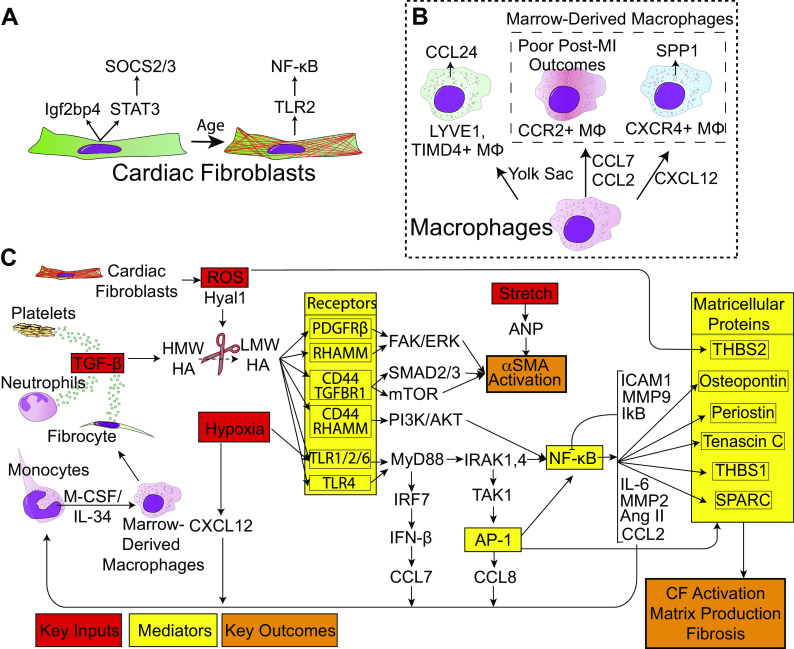
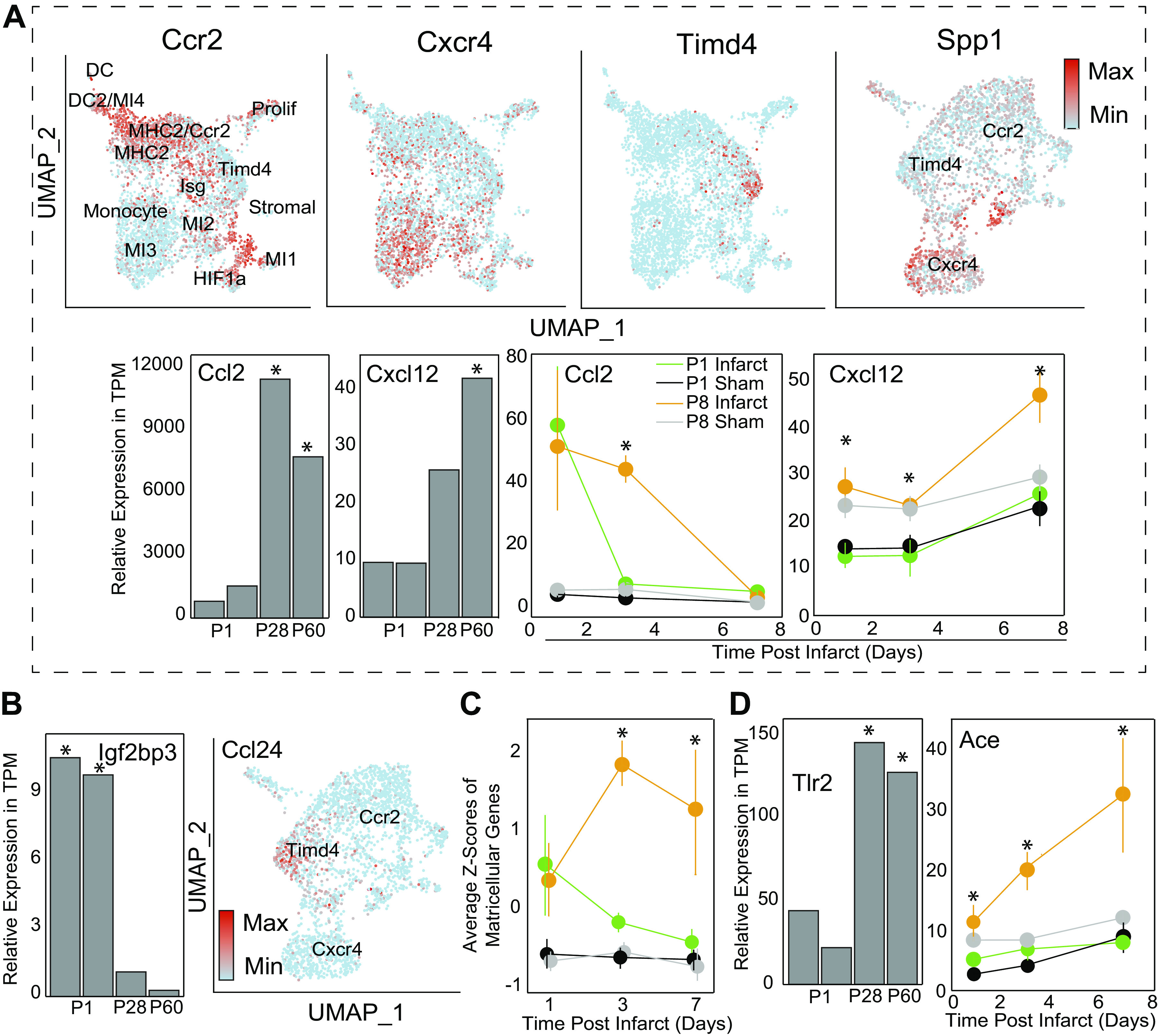
Aside from the first week postnatal, murine heart regeneration is restricted and responses to damage follow classic fibrotic remodeling. Recent transcriptomic analyses have suggested that significant cross talk with the sterile immune response could maintain a more embryonic-like signaling network that promotes acute, transient responses. However, with age, this response-likely mediated by neonatal yolk sac macrophages-then transitions to classical macrophage-mediated, cardiac fibroblast (CF)-based remodeling of the extracellular matrix (ECM) after myocardial infarction (MI). The molecular mechanisms that govern the change with age and drive fibrosis via inflammation are poorly understood. Using multiple ribonucleic acid sequencing (RNA-Seq) datasets, we attempt to resolve the relative contributions of CFs and macrophages in the bulk-healing response of regenerative (postnatal day 1) and nonregenerative hearts (postnatal day 8+). We performed an analysis of bulk RNA-Seq datasets from myocardium and cardiac fibroblasts as well as a single-cell RNA-Seq dataset from cardiac macrophages. MI-specific pathway differences revealed that nonregenerative hearts generated more ECM and had larger matricellular responses correlating with inflammation, produced greater chemotactic gradients to recruit macrophages, and expressed receptors for danger-associated molecular patterns at higher levels than neonates. These changes could result in elevated stress-response pathways compared with neonates, converging at NF-κB and activator protein-1 (AP-1) signaling. Profibrotic gene programs, which greatly diverge on day 3 post MI, lay the foundation for chronic fibrosis, and thus postnatal hearts older than 7 days typically exhibit significantly less regeneration. Our analyses suggest that the macrophage ontogenetic shift in the heart postnatally could result in detrimental stress signaling that suppresses regeneration.NEW & NOTEWORTHY Immediately postnatal mammalian hearts are able to regenerate after infarction, but the cells, pathways, and molecules that regulate this behavior are unclear. By comparing RNA-Seq datasets from regenerative mouse hearts and older, nonregenerative hearts, we are able to identify biological processes that are hallmarks of regeneration. We find that sterile inflammatory processes are upregulated in nonregenerative hearts, initiating profibrotic gene programs 3 days after myocardial infarction that can cause myocardial disease.
Keywords: fibrosis; gene expression and regulation; inflammation; myocardial infarction; myocardial regeneration.
Conflict of interest statement
No conflicts of interest, financial or otherwise, are declared by the authors.
Figures
Comment in
-
Find the stimulus, save the heart: a heroes' story.Am J Physiol Heart Circ Physiol. 2021 Jun 1;320(6):H2185-H2187. doi: 10.1152/ajpheart.00194.2021. Epub 2021 Apr 23. Am J Physiol Heart Circ Physiol. 2021. PMID: 33891514 Free PMC article. No abstract available.
References
-
- King KR, Aguirre AD, Ye YX, Sun Y, Roh JD, Ng RP, Kohler RH, Arlauckas SP, Yoshiko V, Savo A, Sadreyev RI, Kelly M, Fitzgibbons TP, Fitzgerald KA, Mitchison T, Libby P, Nahrendorf M, Weissleder R. IRF3 and type i interferons fuel a fatal response to myocardial infarction. Nat Med 23: 1481–1487, 2017. doi: 10.1038/nm.4428. - DOI - PMC - PubMed
-
- Dick SA, Macklin JA, Nejat S, Momen A, Clemente-Casares X, Althagafi MG, Chen J, Kantores C, Hosseinzadeh S, Aronoff L, Wong A, Zaman R, Barbu I, Besla R, Lavine KJ, Razani B, Ginhoux F, Husain M, Cybulsky MI, Robbins CS, Epalman S. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat Immunol 20: 29–39, 2019. doi: 10.1038/s41590-018-0272-2. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials