

Low-coverage sequencing cost-effectively detects known and novel variation in underrepresented populations
- PMID: 33770507
- PMCID: PMC8059370
- DOI: 10.1016/j.ajhg.2021.03.012
Low-coverage sequencing cost-effectively detects known and novel variation in underrepresented populations
Abstract
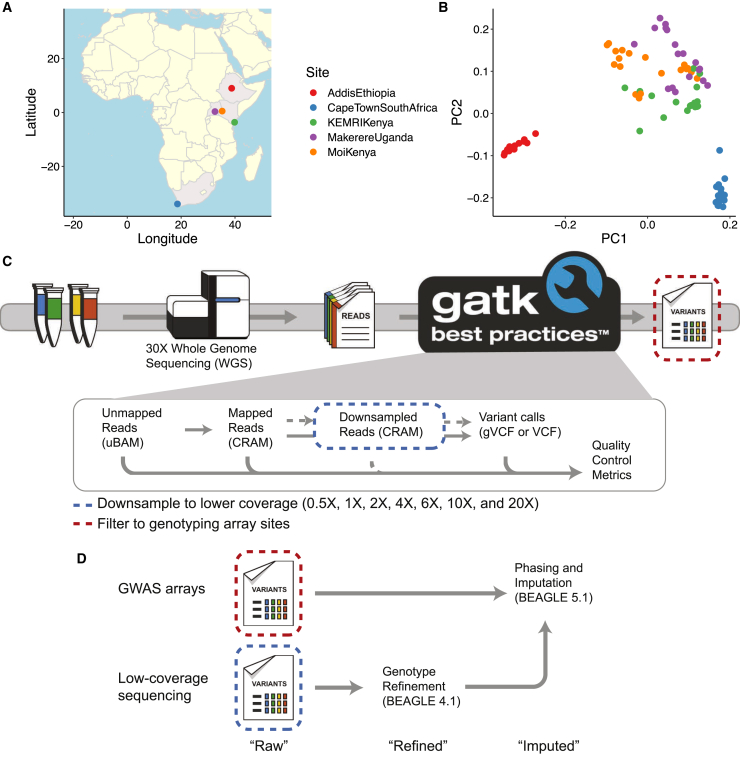
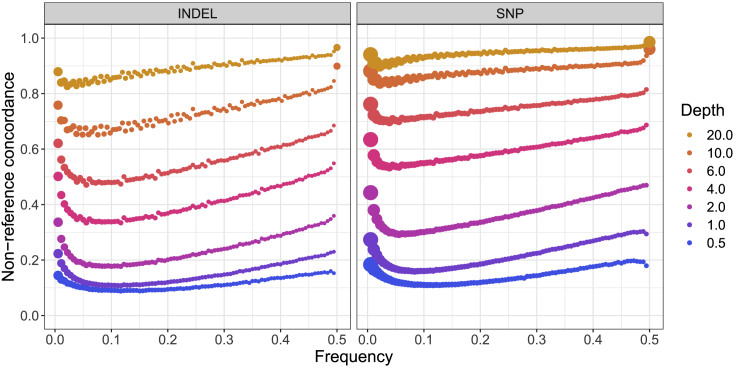
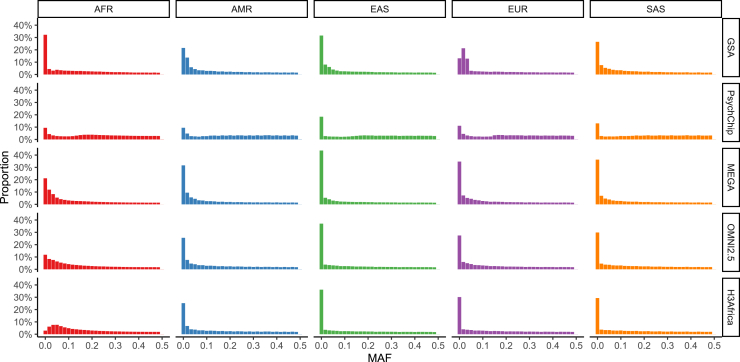
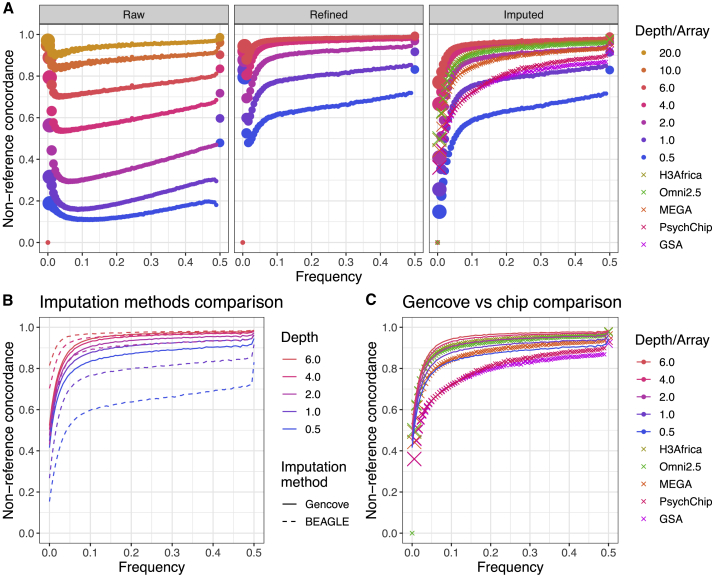
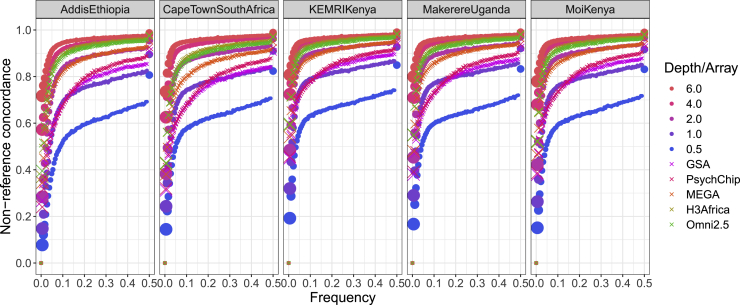
Genetic studies in underrepresented populations identify disproportionate numbers of novel associations. However, most genetic studies use genotyping arrays and sequenced reference panels that best capture variation most common in European ancestry populations. To compare data generation strategies best suited for underrepresented populations, we sequenced the whole genomes of 91 individuals to high coverage as part of the Neuropsychiatric Genetics of African Population-Psychosis (NeuroGAP-Psychosis) study with participants from Ethiopia, Kenya, South Africa, and Uganda. We used a downsampling approach to evaluate the quality of two cost-effective data generation strategies, GWAS arrays versus low-coverage sequencing, by calculating the concordance of imputed variants from these technologies with those from deep whole-genome sequencing data. We show that low-coverage sequencing at a depth of ≥4× captures variants of all frequencies more accurately than all commonly used GWAS arrays investigated and at a comparable cost. Lower depths of sequencing (0.5-1×) performed comparably to commonly used low-density GWAS arrays. Low-coverage sequencing is also sensitive to novel variation; 4× sequencing detects 45% of singletons and 95% of common variants identified in high-coverage African whole genomes. Low-coverage sequencing approaches surmount the problems induced by the ascertainment of common genotyping arrays, effectively identify novel variation particularly in underrepresented populations, and present opportunities to enhance variant discovery at a cost similar to traditional approaches.
Keywords: Africa; GWAS; GWAS arrays; cost comparison; low-coverage sequencing; study design; whole-genome sequencing.
Copyright © 2021 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
A.R.M. has consulted for 23andMe and Illumina. B.M.N. is a member of the Deep Genomics Scientific Advisory Board. He also serves as a consultant for the Camp4 Therapeutics Corporation, Takeda Pharmaceutical, and Biogen. M.J.D. is a founder of Maze Therapeutics. J.K.P. is an employee of Gencove, Inc. D.J.S. has received research grants and/or consultancy honoraria from Lundbeck and Sun. The remaining authors declare no competing interests.
Figures
References
-
- Marchini J., Howie B. Genotype imputation for genome-wide association studies. Nat. Rev. Genet. 2010;11:499–511. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
