A tumor-specific mechanism of Treg enrichment mediated by the integrin αvβ8
- PMID: 33771888
- PMCID: PMC8425767
- DOI: 10.1126/sciimmunol.abf0558
A tumor-specific mechanism of Treg enrichment mediated by the integrin αvβ8
Abstract
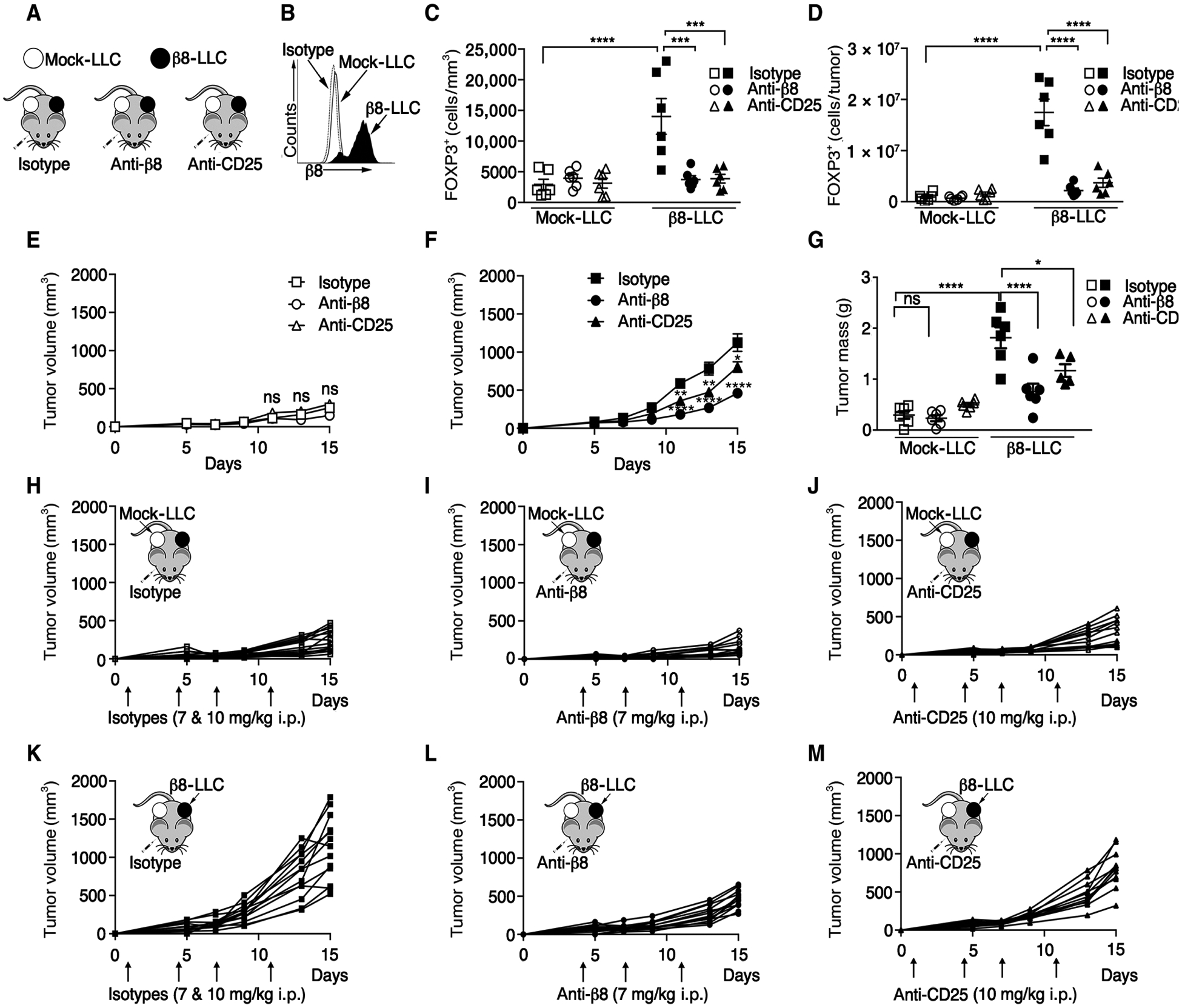
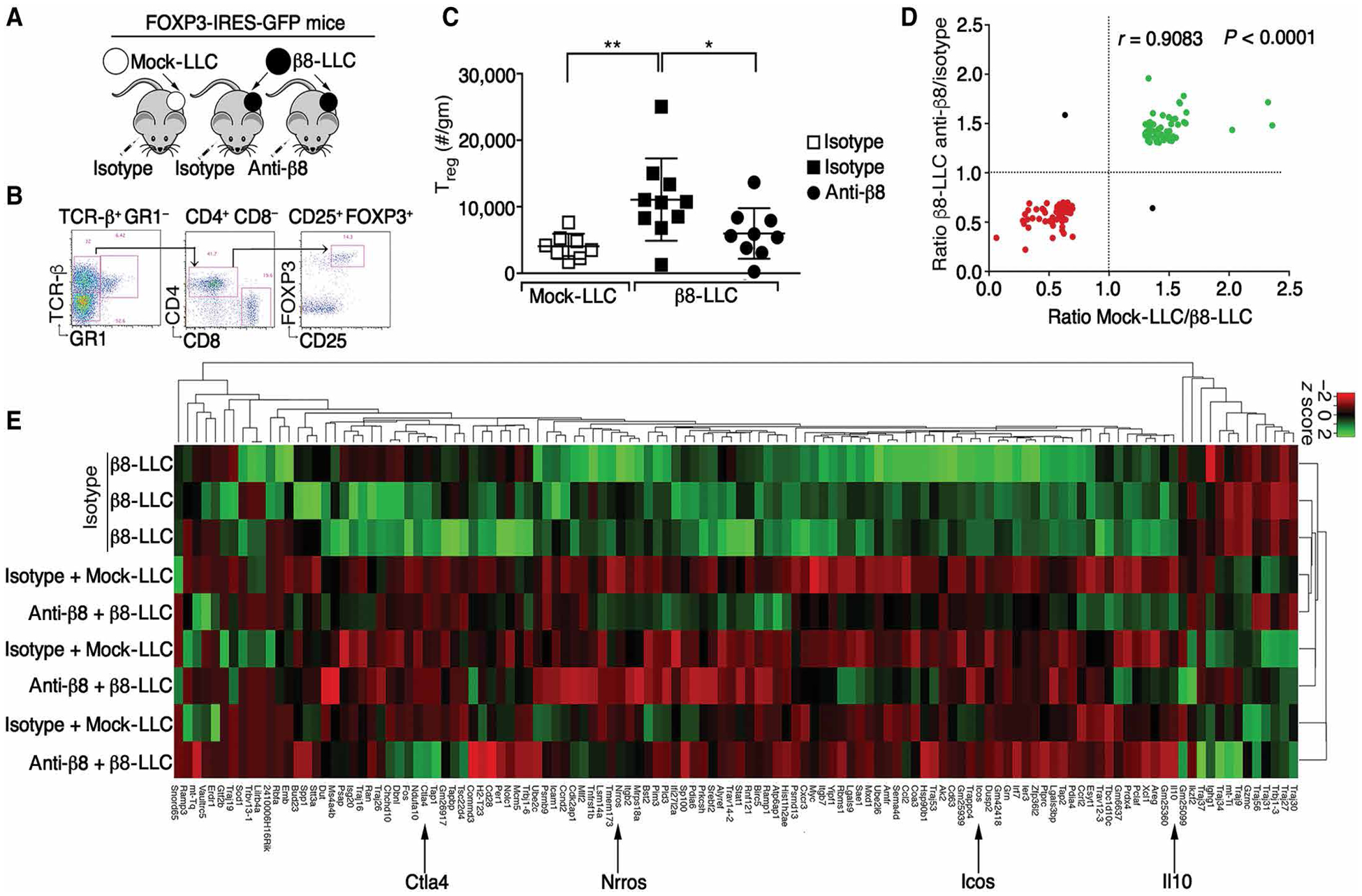
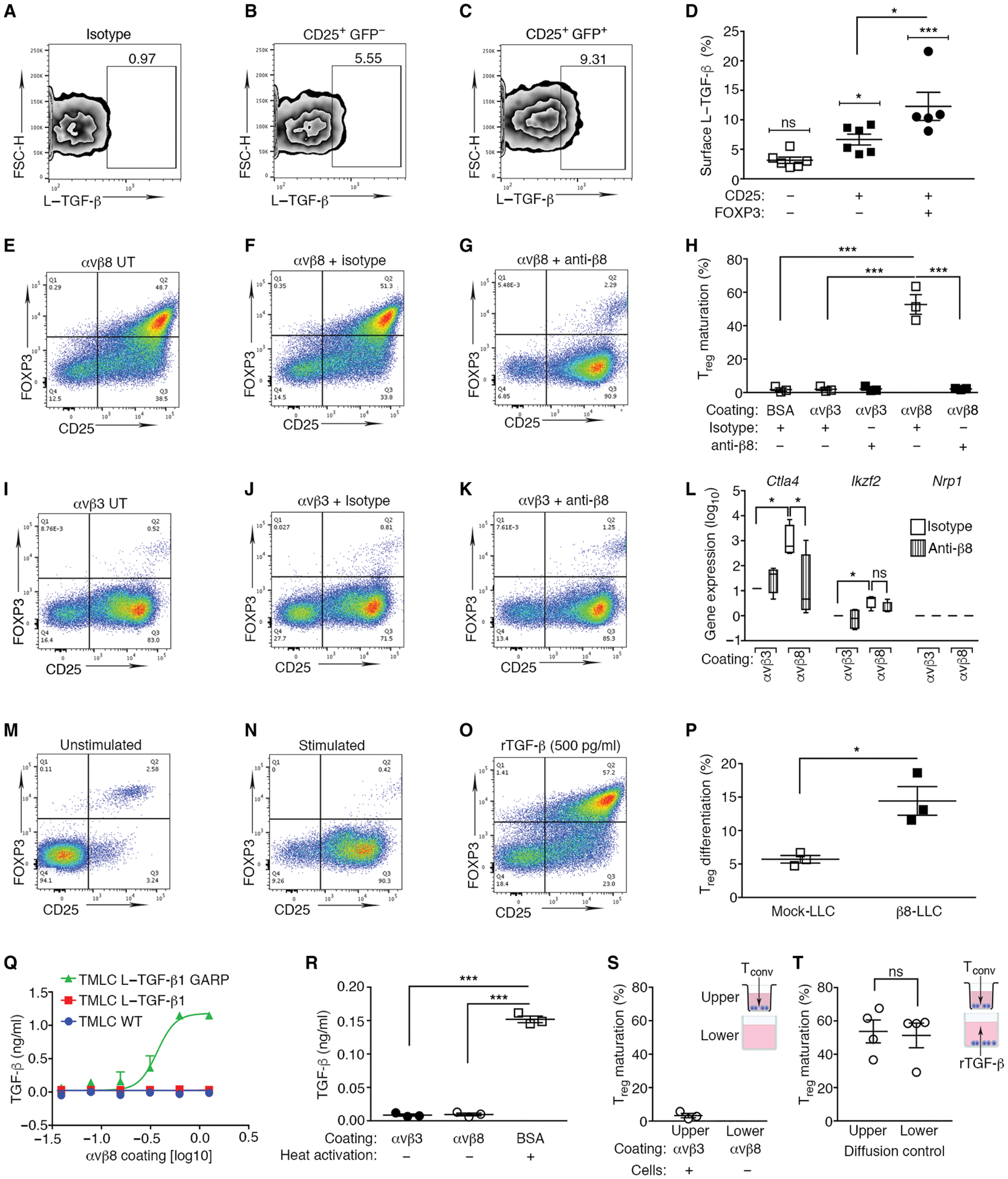
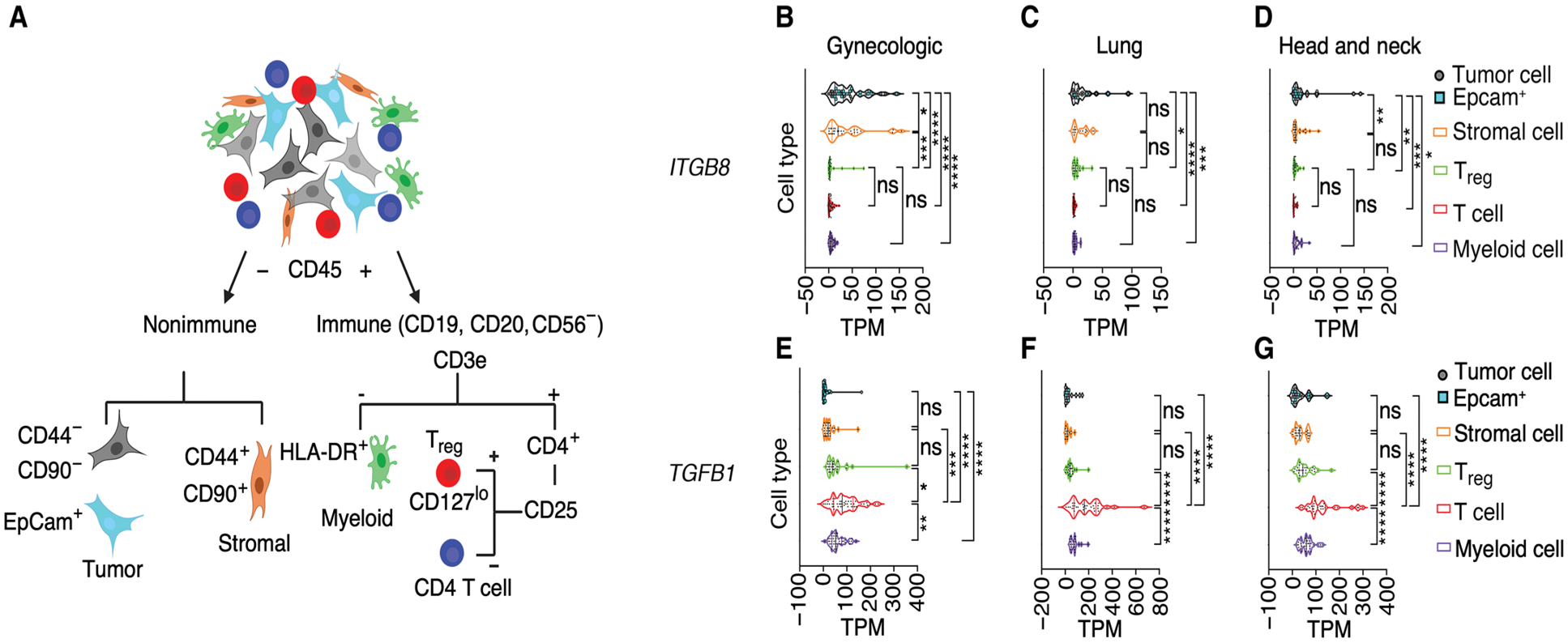
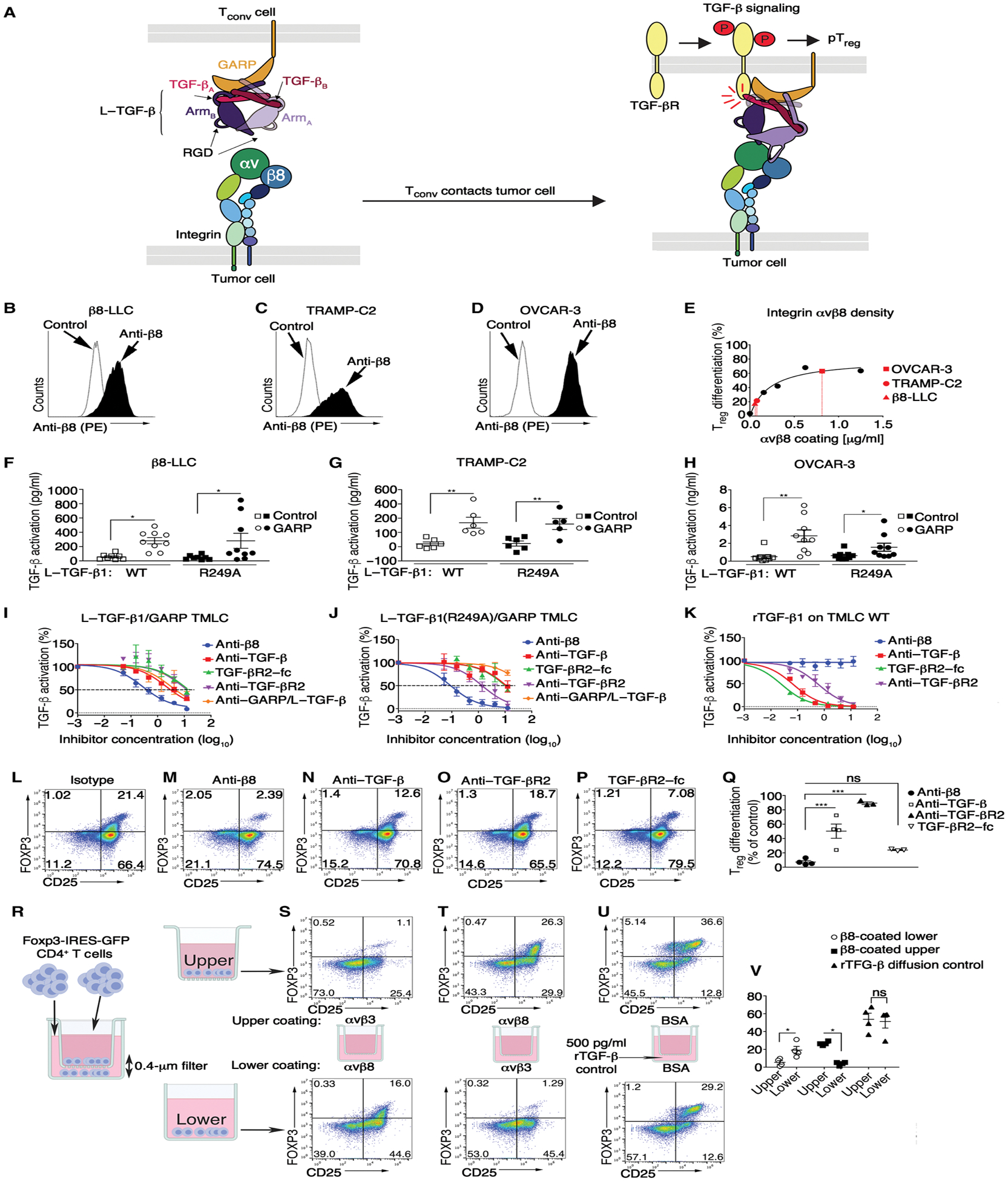
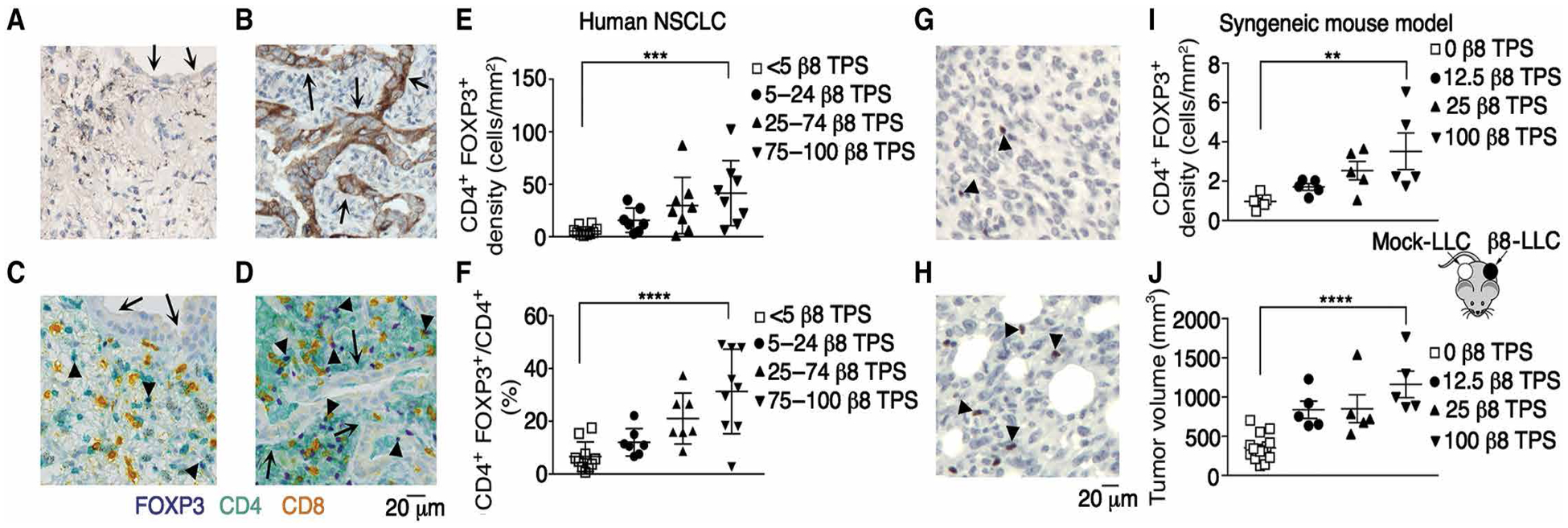
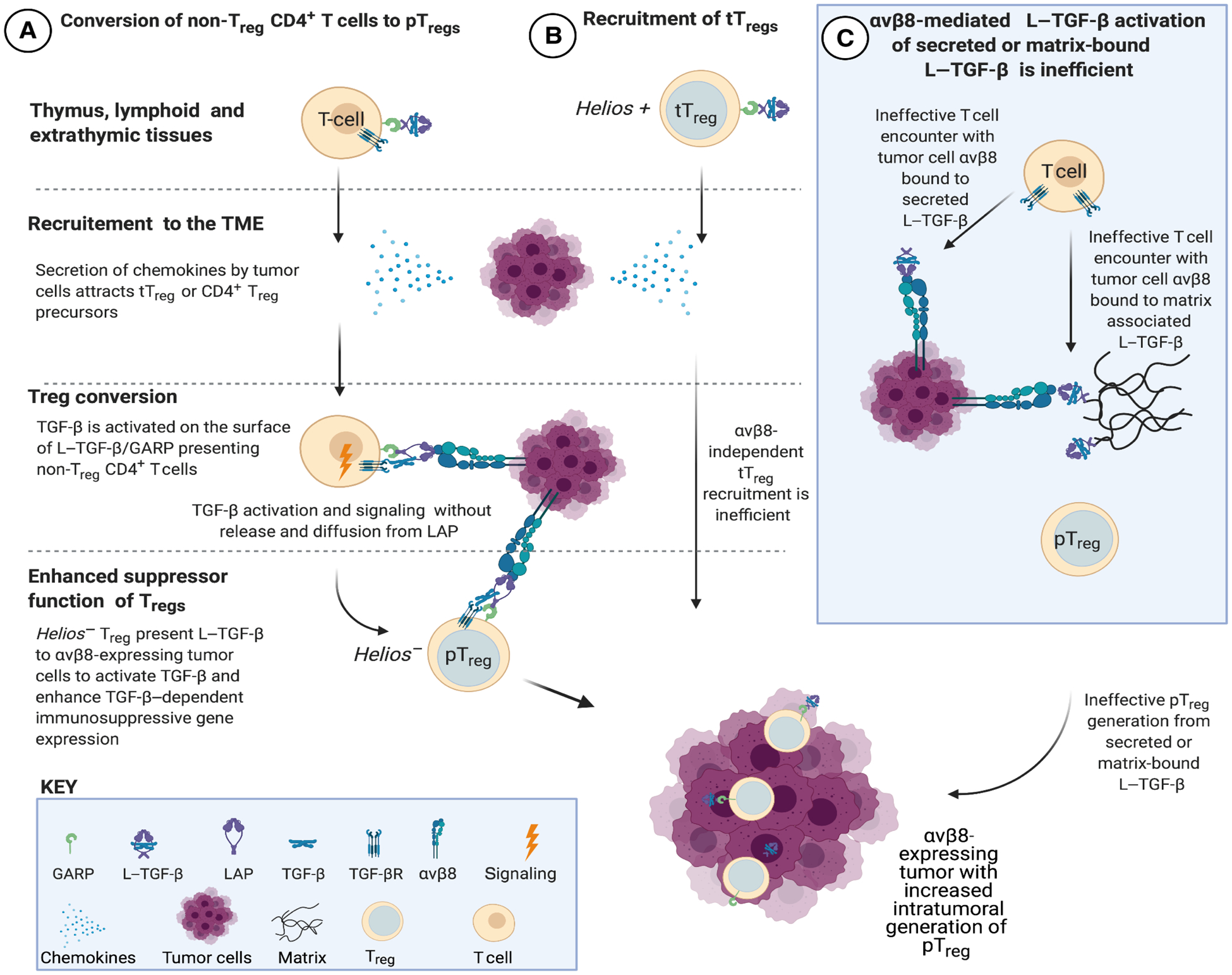
Regulatory T cells (Tregs) that promote tumor immune evasion are enriched in certain tumors and correlate with poor prognosis. However, mechanisms for Treg enrichment remain incompletely understood. We described a mechanism for Treg enrichment in mouse and human tumors mediated by the αvβ8 integrin. Tumor cell αvβ8 bound to latent transforming growth factor-β (L-TGF-β) presented on the surface of T cells, resulting in TGF-β activation and immunosuppressive Treg differentiation in vitro. In vivo, tumor cell αvβ8 expression correlated with Treg enrichment, immunosuppressive Treg gene expression, and increased tumor growth, which was reduced in mice by αvβ8 inhibition or Treg depletion. Structural modeling and cell-based studies suggested a highly geometrically constrained complex forming between αvβ8-expressing tumor cells and L-TGF-β-expressing T cells, facilitating TGF-β activation, independent of release and diffusion, and providing limited access to TGF-β inhibitors. These findings suggest a highly localized tumor-specific mechanism for Treg enrichment.
Copyright © 2021 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Figures
References
-
- Fridman WH, Pagès F, Sautès-Fridman C, Galon J, The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 12, 298–306 (2012). - PubMed
-
- Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zhu Y, Wei S, Kryczek I, Daniel B, Gordon A, Myers L, Lackner A, Disis ML, Knutson KL, Chen L, Zou W, Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med 10, 942–949 (2004). - PubMed
-
- Shimizu J, Yamazaki S, Sakaguchi S, Induction of tumor immunity by removing CD25+CD4+ T cells: A common basis between tumor immunity and autoimmunity. J. Immunol 163, 5211–5218 (1999). - PubMed
-
- Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E, Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor α) monoclonal antibody. Cancer Res. 59, 3128–3133 (1999). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- U54 HL119893/HL/NHLBI NIH HHS/United States
- K01 DK099405/DK/NIDDK NIH HHS/United States
- T32 AI007334/AI/NIAID NIH HHS/United States
- P30 DK026743/DK/NIDDK NIH HHS/United States
- R01 HL113032/HL/NHLBI NIH HHS/United States
- F31 DK112607/DK/NIDDK NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- S10 OD020054/OD/NIH HHS/United States
- U01 CA217864/CA/NCI NIH HHS/United States
- P41 CA196276/CA/NCI NIH HHS/United States
- R01 HL165175/HL/NHLBI NIH HHS/United States
- R01 CA197363/CA/NCI NIH HHS/United States
- R01 DK093646/DK/NIDDK NIH HHS/United States
- R01 HL134183/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials