

Cardiac macrophages prevent sudden death during heart stress
- PMID: 33771995
- PMCID: PMC7997915
- DOI: 10.1038/s41467-021-22178-0
Cardiac macrophages prevent sudden death during heart stress
Abstract
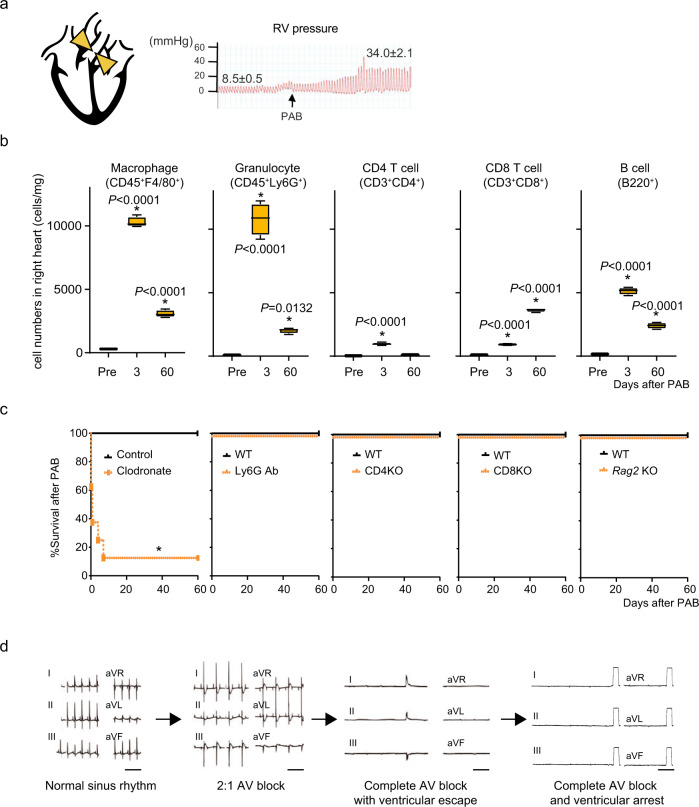
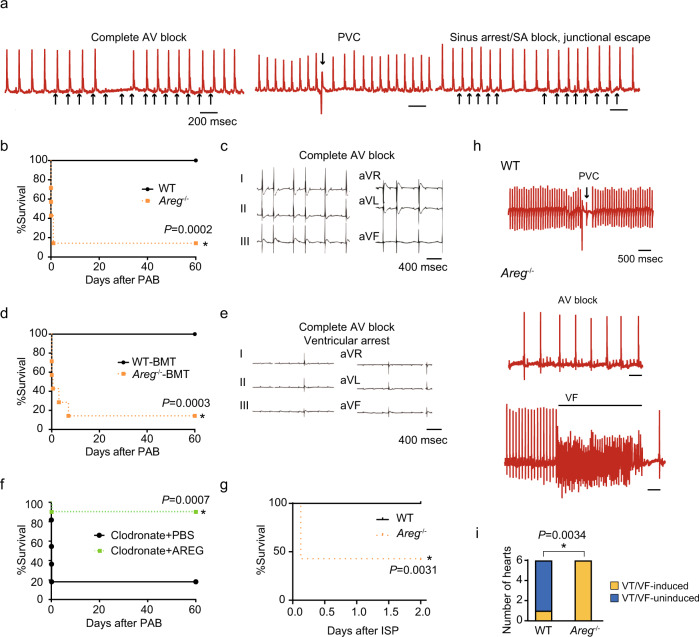
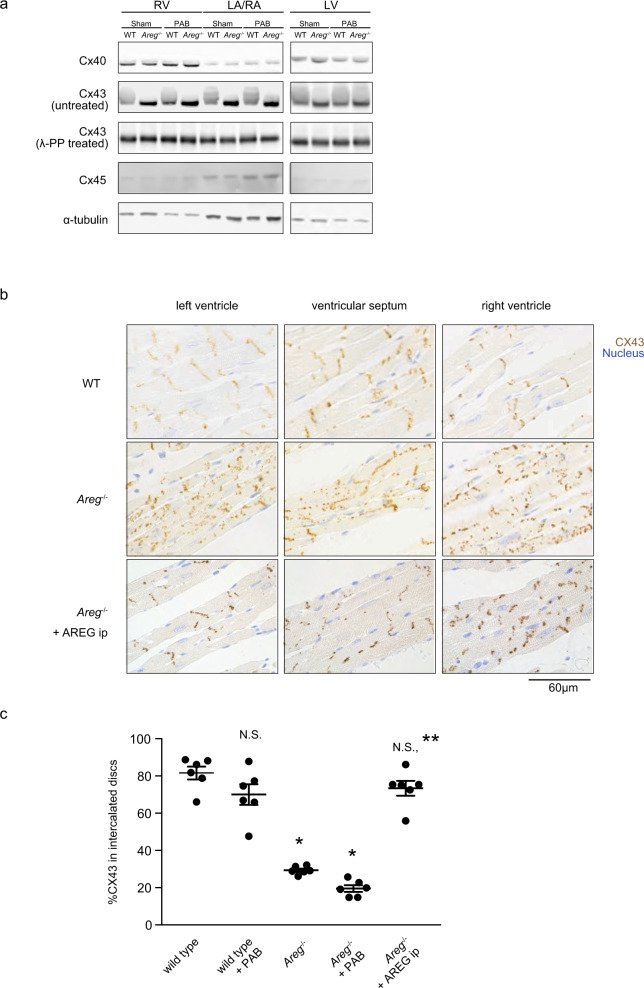
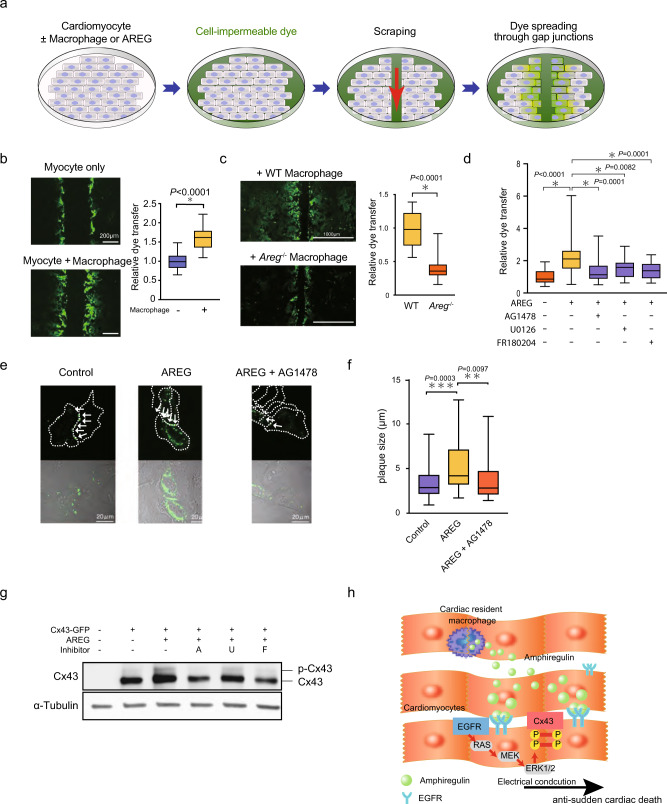
Cardiac arrhythmias are a primary contributor to sudden cardiac death, a major unmet medical need. Because right ventricular (RV) dysfunction increases the risk for sudden cardiac death, we examined responses to RV stress in mice. Among immune cells accumulated in the RV after pressure overload-induced by pulmonary artery banding, interfering with macrophages caused sudden death from severe arrhythmias. We show that cardiac macrophages crucially maintain cardiac impulse conduction by facilitating myocardial intercellular communication through gap junctions. Amphiregulin (AREG) produced by cardiac macrophages is a key mediator that controls connexin 43 phosphorylation and translocation in cardiomyocytes. Deletion of Areg from macrophages led to disorganization of gap junctions and, in turn, lethal arrhythmias during acute stresses, including RV pressure overload and β-adrenergic receptor stimulation. These results suggest that AREG from cardiac resident macrophages is a critical regulator of cardiac impulse conduction and may be a useful therapeutic target for the prevention of sudden death.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Naksuk N, et al. Right ventricular dysfunction and long-term risk of sudden cardiac death in patients with and without severe left ventricular dysfunction. Circulation: Arrhythmia Electrophysiol. 2018;11:e006091. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
