

Italian cohort of Lafora disease: Clinical features, disease evolution, and genotype-phenotype correlations
- PMID: 33773408
- PMCID: PMC8166462
- DOI: 10.1016/j.jns.2021.117409
Italian cohort of Lafora disease: Clinical features, disease evolution, and genotype-phenotype correlations
Abstract
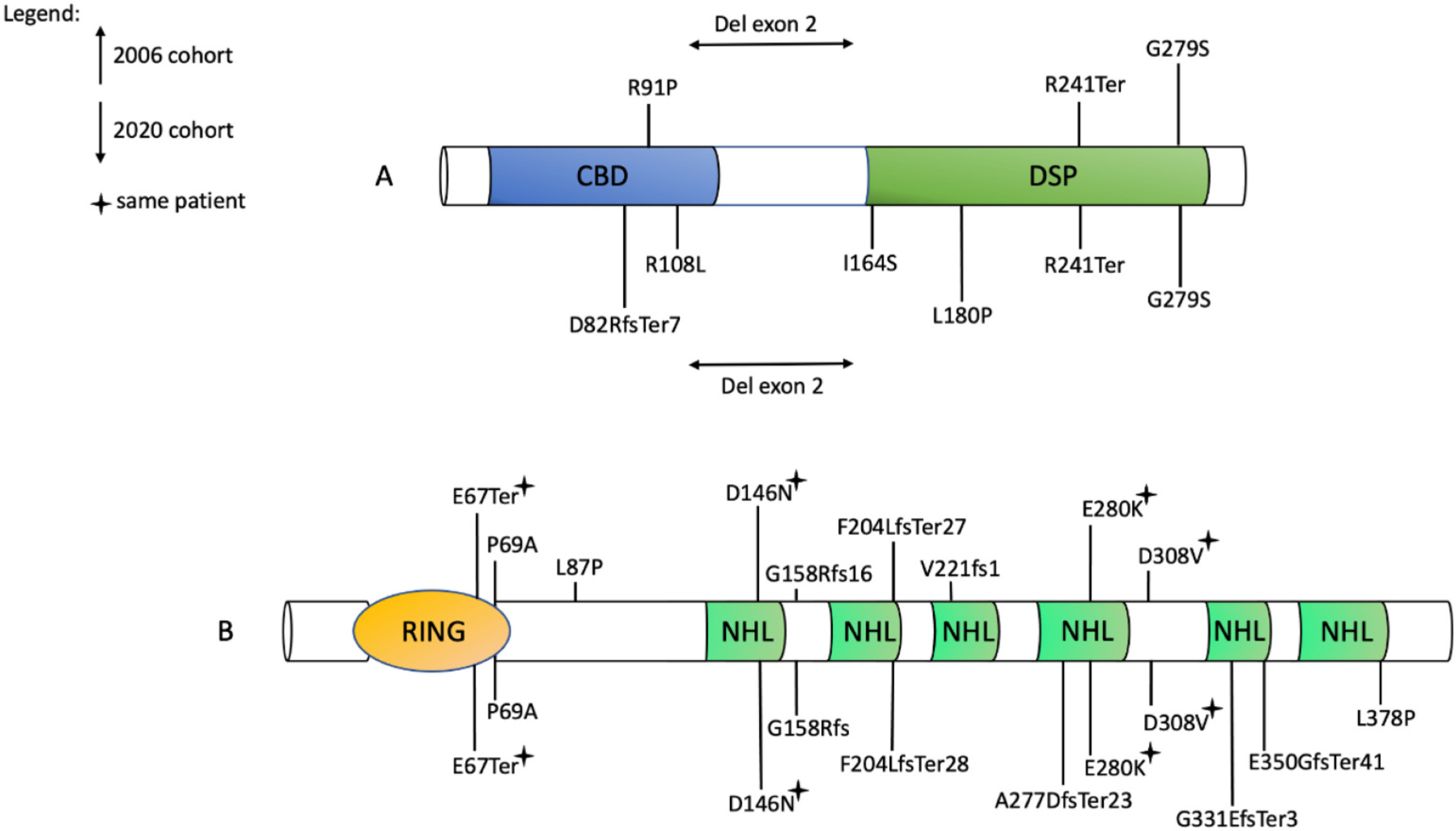
Background: Lafora disease (LD) is characterized by progressive myoclonus, refractory epilepsy, and cognitive deterioration. This complex neurodegenerative condition is caused by pathogenic variants in EPM2A/EPM2B genes, encoding two essential glycogen metabolism enzymes known as laforin and malin. Long-term follow-up data are lacking. We describe the clinical features and genetic findings of a cohort of 26 Italian patients with a long clinical follow-up.
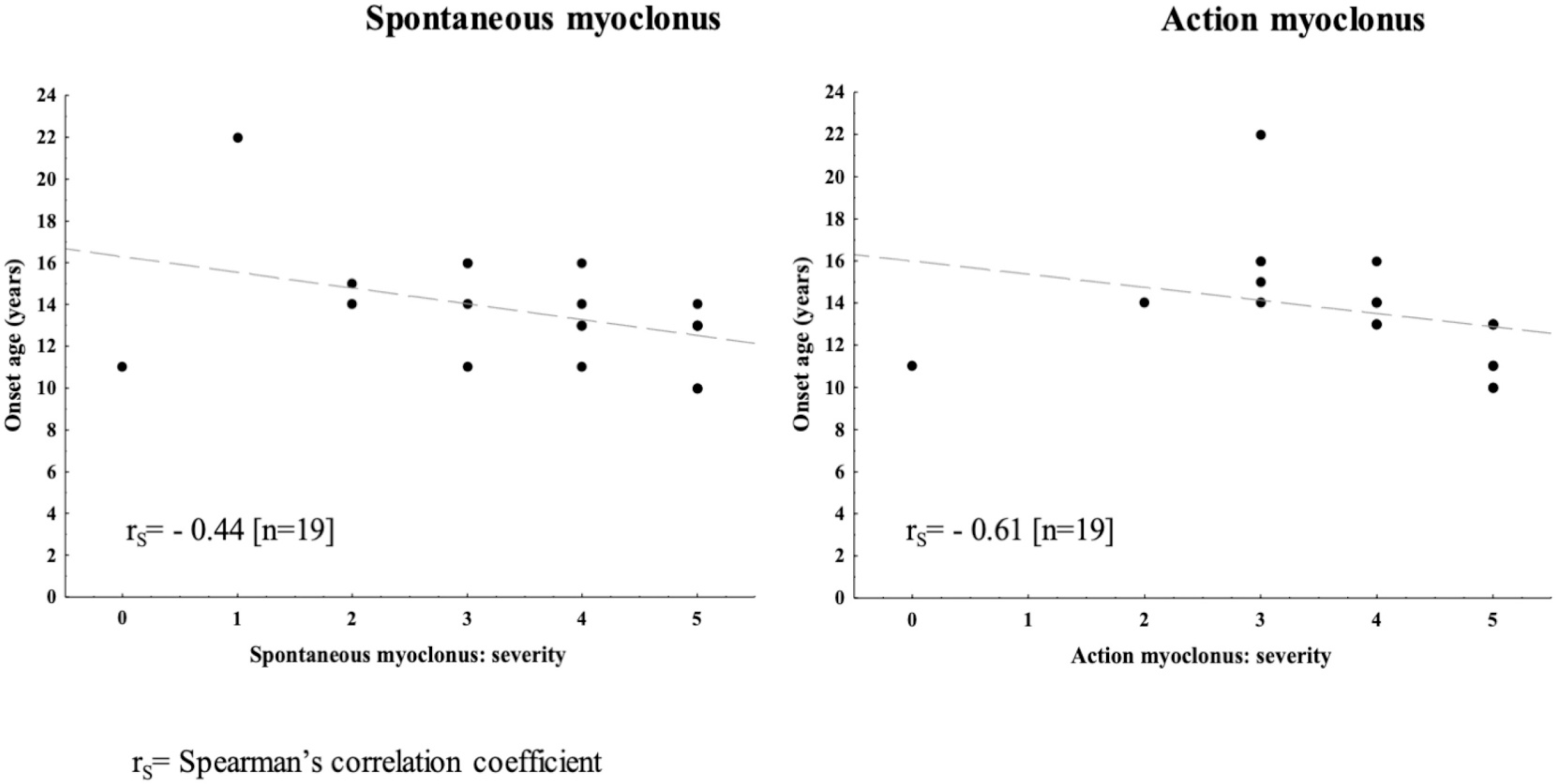
Methods: Patients with EPM2A/EPM2B pathogenic variants were identified by direct gene sequencing or gene panels with targeted re-sequencing. Disease progression, motor functions, and mental performance were assessed by a simplified disability scale. Spontaneous/action myoclonus severity was scored by the Magaudda Scale.
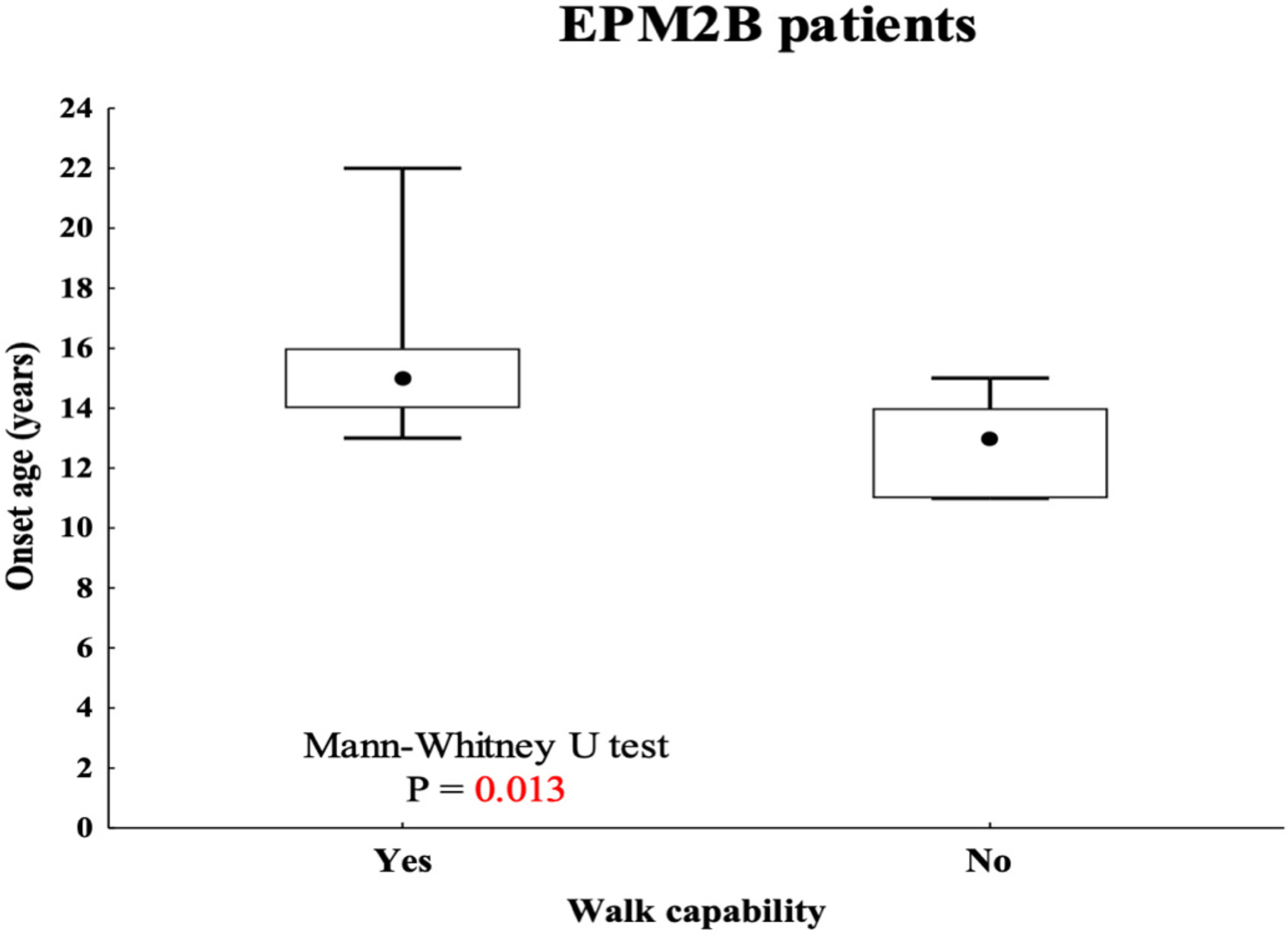
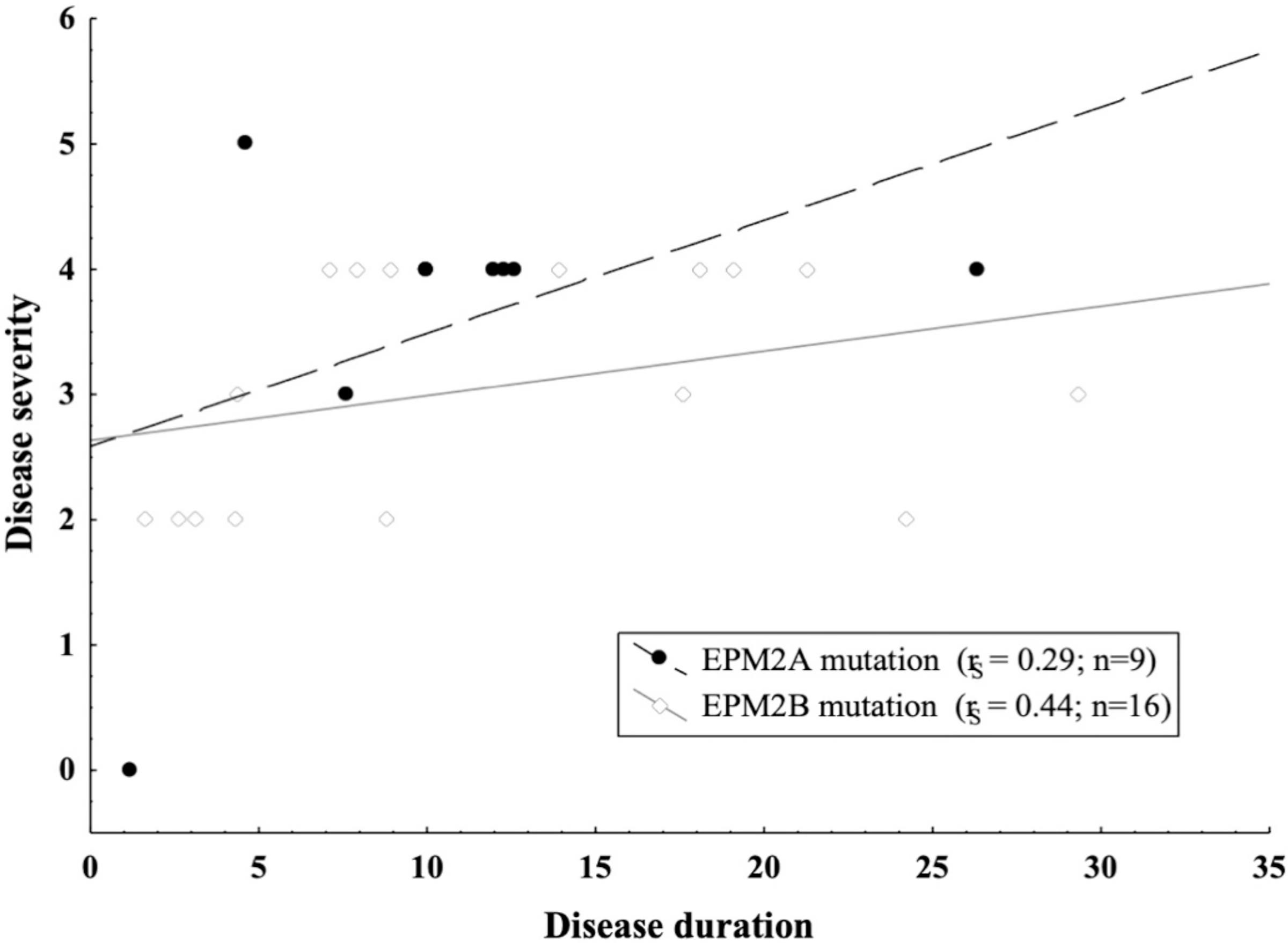
Results: Age range was 12.2-46.2 years (mean:25.53 ± 9.14). Age at disease onset ranged from 10 to 22 years (mean:14.04 ± 2.62). The mean follow-up period was 11.48 ± 7.8 years. Twelve out of the 26 (46%) patients preserved walking ability and 13 (50%) maintained speech. A slower disease progression with preserved ambulation and speech after ≥4 years of follow-up was observed in 1 (11%) out of the 9 (35%) EPM2A patients and in 6 (35%) out of the 17 (65%) EPM2B patients. Follow-up was >10 years in 7 (41.2%) EPM2B individuals, including two harbouring the homozygous p.(D146N) pathogenic variant.
Conclusions: This study supports an overall worse disease outcome with severe deterioration of ambulation and speech in patients carrying EPM2A mutations. However, the delayed onset of disabling symptoms observed in the EPM2B subjects harbouring the p.(D146N) pathogenic variant suggests that the underlying causative variant may still influence LD severity.
Keywords: EPM2A; EPM2B; Epilepsy; Lafora disease; Neurodegeneration; Progressive myoclonus.
Copyright © 2021 Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of Competing Interest
P.S. has received speaker fees and participated at advisory boards for Biomarin, Zogenyx, GW Pharmaceuticals, and has received research funding by ENECTA BV, GW Pharmaceuticals, Kolfarma srl., Eisai. The other authors do not report any conflict of interest.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
