

Phylogenomic analysis of COVID-19 summer and winter outbreaks in Hong Kong: An observational study
- PMID: 33778795
- PMCID: PMC7985010
- DOI: 10.1016/j.lanwpc.2021.100130
Phylogenomic analysis of COVID-19 summer and winter outbreaks in Hong Kong: An observational study
Abstract
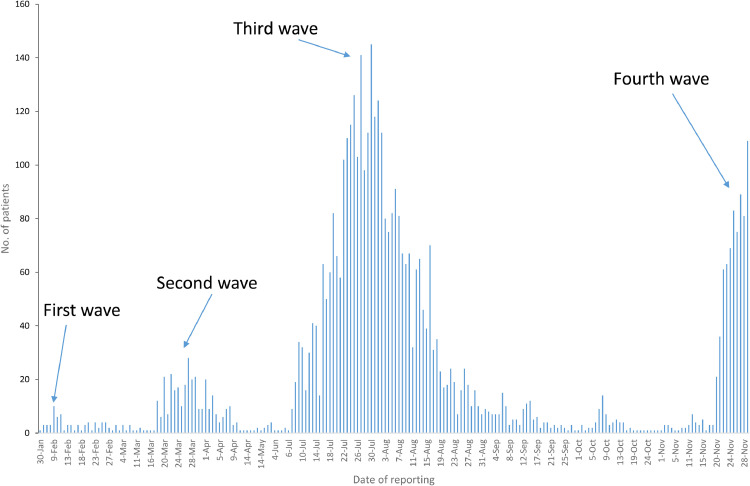
Background: Viral genomic surveillance is vital for understanding the transmission of COVID-19. In Hong Kong, breakthrough outbreaks have occurred in July (third wave) and November (fourth wave) 2020. We used whole viral genome analysis to study the characteristics of these waves.
Methods: We analyzed 509 SARS-CoV-2 genomes collected from Hong Kong patients between 22nd January and 29th November, 2020. Phylogenetic and phylodynamic analyses were performed, and were interpreted with epidemiological information.
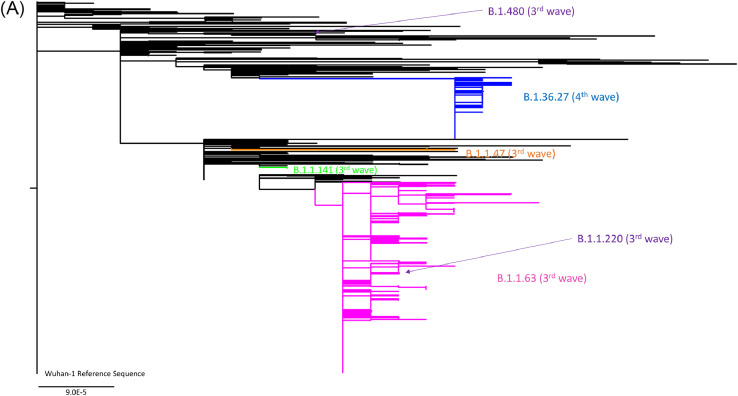
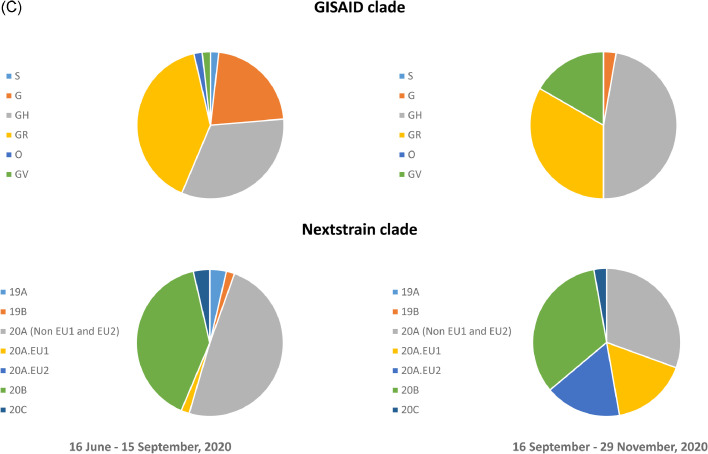
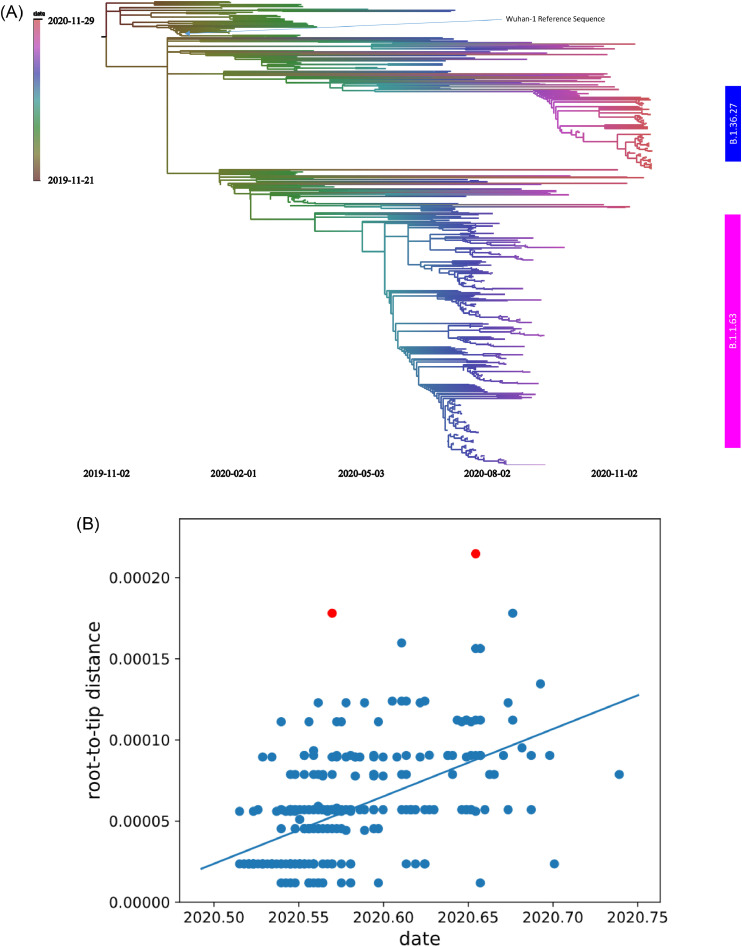
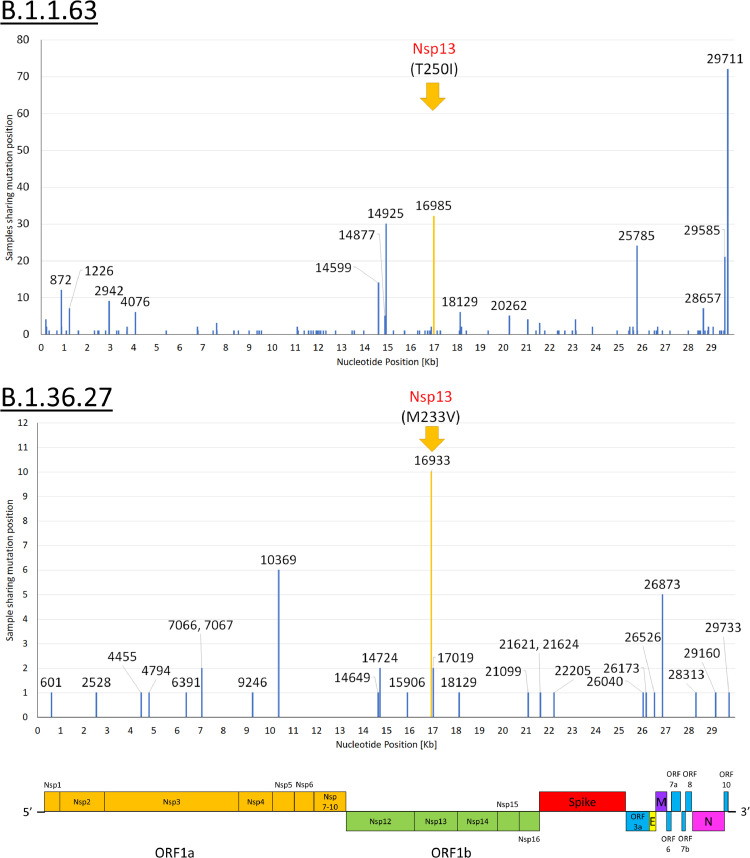
Findings: During the third and fourth waves, diverse SARS-CoV-2 genomes were identified among imported infections. Conversely, local infections were dominated by a single lineage during each wave, with 96.6% (259/268) in the third wave and 100% (73/73) in the fourth wave belonging to B.1.1.63 and B.1.36.27 lineages, respectively. While B.1.1.63 lineage was imported 2 weeks before the beginning of the third wave, B.1.36.27 lineage has circulated in Hong Kong for 2 months prior to the fourth wave. During the fourth wave, 50.7% (37/73) of local infections in November was identical to the viral genome from an imported case in September. Within B.1.1.63 or B.1.36.27 lineage in our cohort, the most common non-synonymous mutations occurred at the helicase (nsp13) gene.
Interpretation: Although stringent measures have prevented most imported cases from spreading in Hong Kong, a single lineage with low-level local transmission in October and early November was responsible for the fourth wave. A superspreading event or lower temperature in November may have facilitated the spread of the B.1.36.27 lineage.
Keywords: COVID19; Next generation sequencing; Outbreak; Phylodynamic; Phylogenetic; SARS-CoV-2; Viral genome.
© 2021 The Author(s). Published by Elsevier Ltd.
Conflict of interest statement
All authors declare no conflict of interest.
Figures

References
-
- Oude Munnink B.B., Nieuwenhuijse D.F., Stein M., O'Toole A., Haverkate M., Mollers M. Rapid SARS-CoV-2 whole-genome sequencing and analysis for informed public health decision-making in the Netherlands. Nat. Med. 2020;26:1405–1410. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
