

Thousands of previously unknown phages discovered in whole-community human gut metagenomes
- PMID: 33781338
- PMCID: PMC8008677
- DOI: 10.1186/s40168-021-01017-w
Thousands of previously unknown phages discovered in whole-community human gut metagenomes
Abstract
Background: Double-stranded DNA bacteriophages (dsDNA phages) play pivotal roles in structuring human gut microbiomes; yet, the gut virome is far from being fully characterized, and additional groups of phages, including highly abundant ones, continue to be discovered by metagenome mining. A multilevel framework for taxonomic classification of viruses was recently adopted, facilitating the classification of phages into evolutionary informative taxonomic units based on hallmark genes. Together with advanced approaches for sequence assembly and powerful methods of sequence analysis, this revised framework offers the opportunity to discover and classify unknown phage taxa in the human gut.
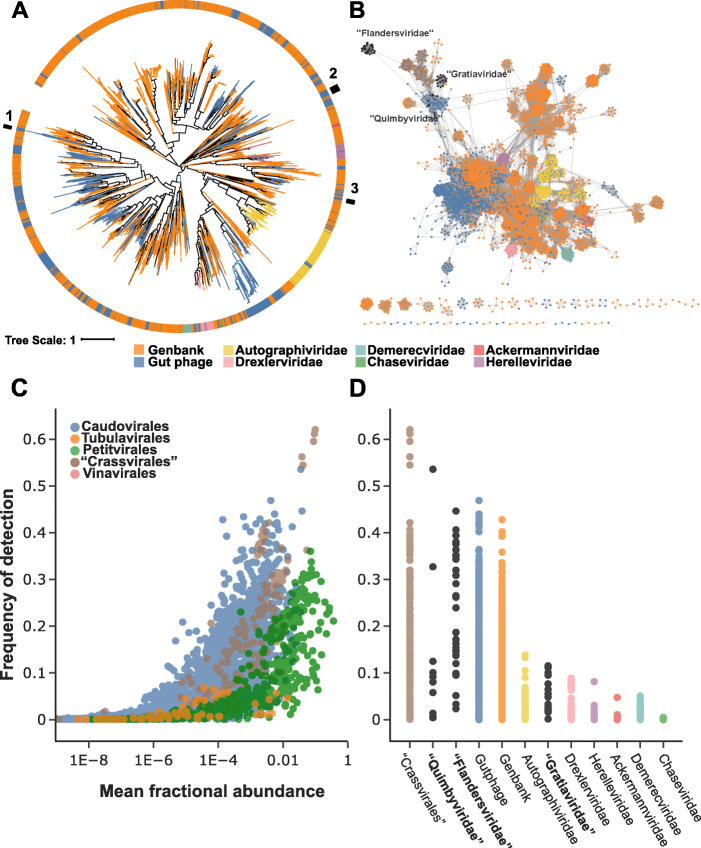
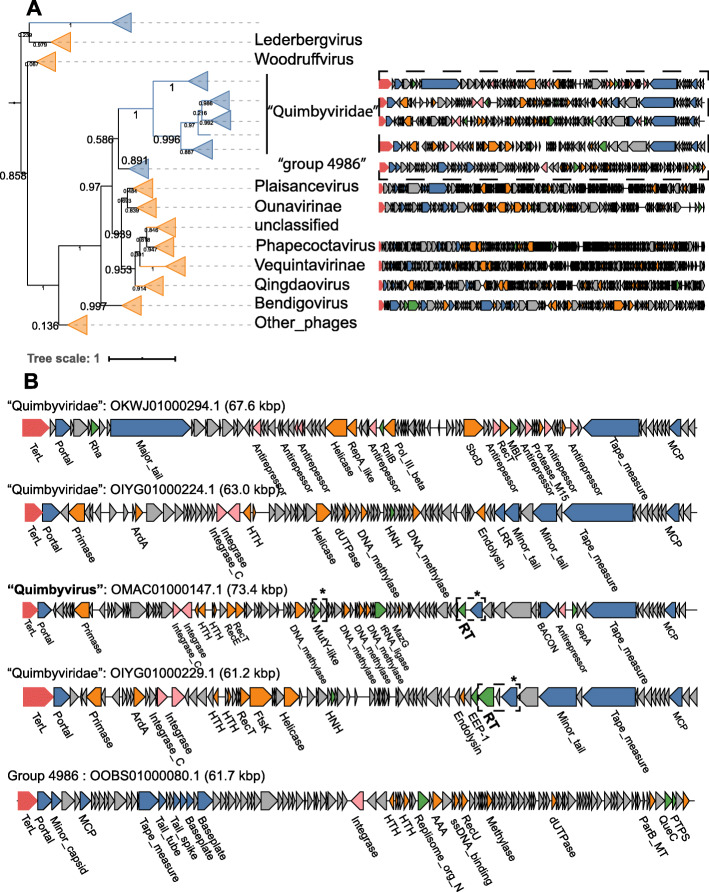
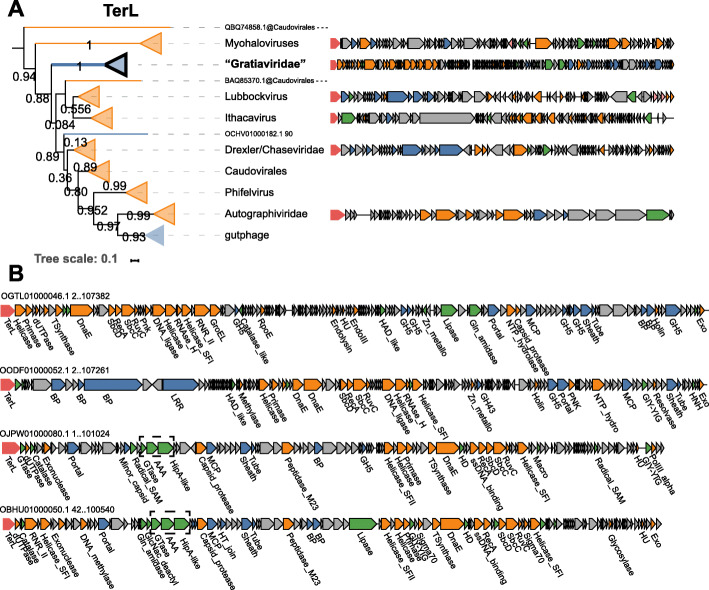
Results: A search of human gut metagenomes for circular contigs encoding phage hallmark genes resulted in the identification of 3738 apparently complete phage genomes that represent 451 putative genera. Several of these phage genera are only distantly related to previously identified phages and are likely to found new families. Two of the candidate families, "Flandersviridae" and "Quimbyviridae", include some of the most common and abundant members of the human gut virome that infect Bacteroides, Parabacteroides, and Prevotella. The third proposed family, "Gratiaviridae," consists of less abundant phages that are distantly related to the families Autographiviridae, Drexlerviridae, and Chaseviridae. Analysis of CRISPR spacers indicates that phages of all three putative families infect bacteria of the phylum Bacteroidetes. Comparative genomic analysis of the three candidate phage families revealed features without precedent in phage genomes. Some "Quimbyviridae" phages possess Diversity-Generating Retroelements (DGRs) that generate hypervariable target genes nested within defense-related genes, whereas the previously known targets of phage-encoded DGRs are structural genes. Several "Flandersviridae" phages encode enzymes of the isoprenoid pathway, a lipid biosynthesis pathway that so far has not been known to be manipulated by phages. The "Gratiaviridae" phages encode a HipA-family protein kinase and glycosyltransferase, suggesting these phages modify the host cell wall, preventing superinfection by other phages. Hundreds of phages in these three and other families are shown to encode catalases and iron-sequestering enzymes that can be predicted to enhance cellular tolerance to reactive oxygen species.
Conclusions: Analysis of phage genomes identified in whole-community human gut metagenomes resulted in the delineation of at least three new candidate families of Caudovirales and revealed diverse putative mechanisms underlying phage-host interactions in the human gut. Addition of these phylogenetically classified, diverse, and distinct phages to public databases will facilitate taxonomic decomposition and functional characterization of human gut viromes. Video abstract.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

References
-
- Barr JJ, Auro R, Furlan M, Whiteson KL, Erb ML, Pogliano J, Stotland A, Wolkowicz R, Cutting AS, Doran KS, Salamon P, Youle M, Rohwer F. Bacteriophage adhering to mucus provide a non–host-derived immunity. Proc Natl Acad Sci. 2013;110(26):10771–10776. doi: 10.1073/pnas.1305923110. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
