

Iduronate-2-sulfatase fused with anti-hTfR antibody, pabinafusp alfa, for MPS-II: A phase 2 trial in Brazil
- PMID: 33781915
- PMCID: PMC8261166
- DOI: 10.1016/j.ymthe.2021.03.019
Iduronate-2-sulfatase fused with anti-hTfR antibody, pabinafusp alfa, for MPS-II: A phase 2 trial in Brazil
Abstract
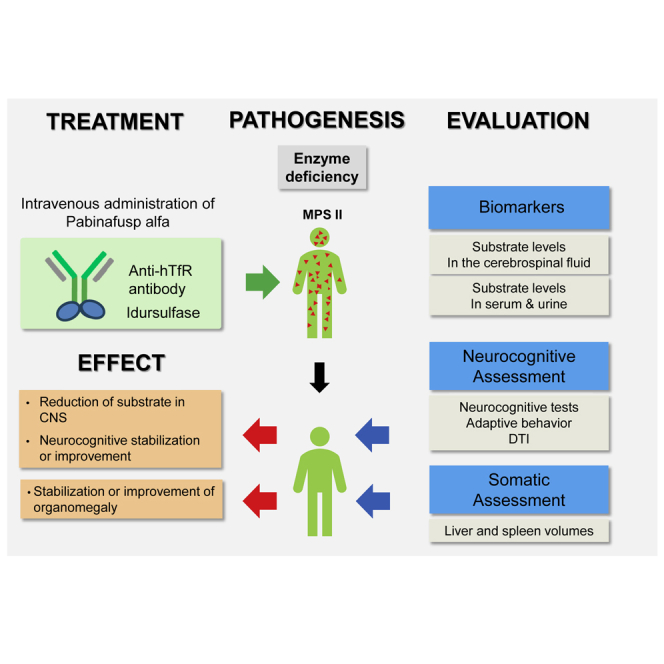
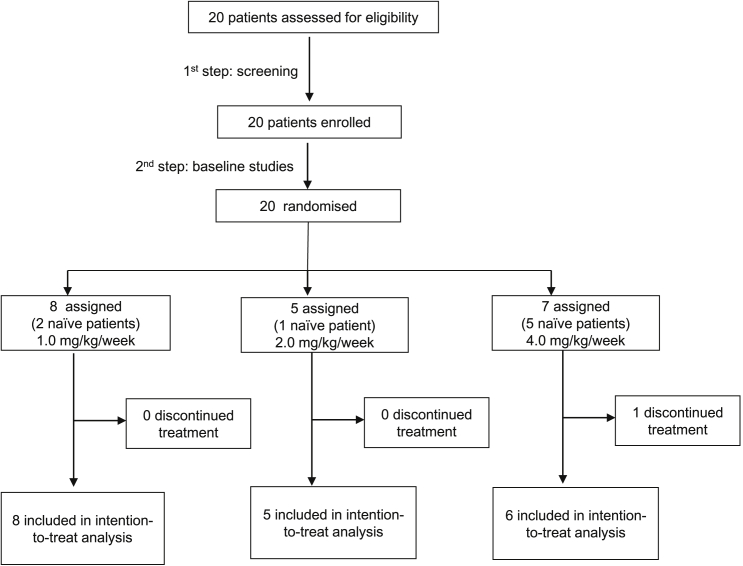
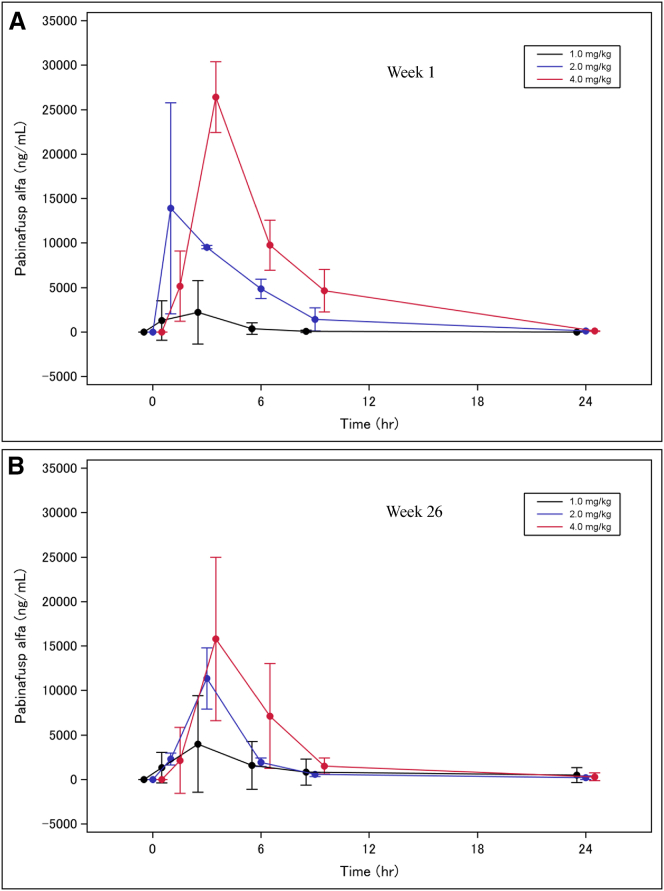
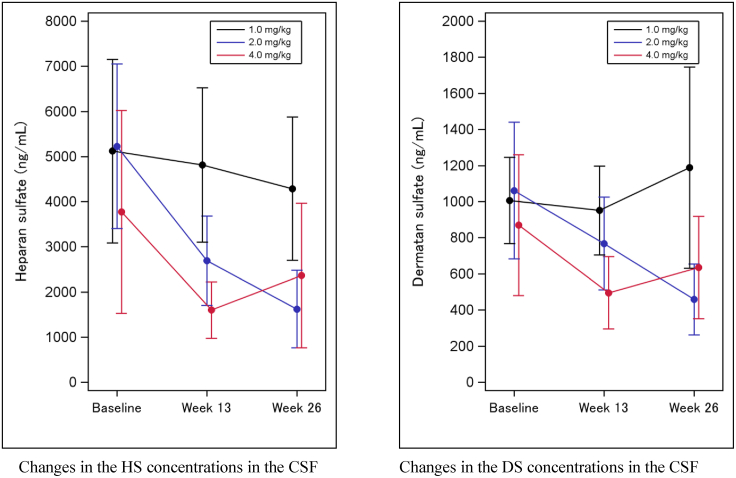
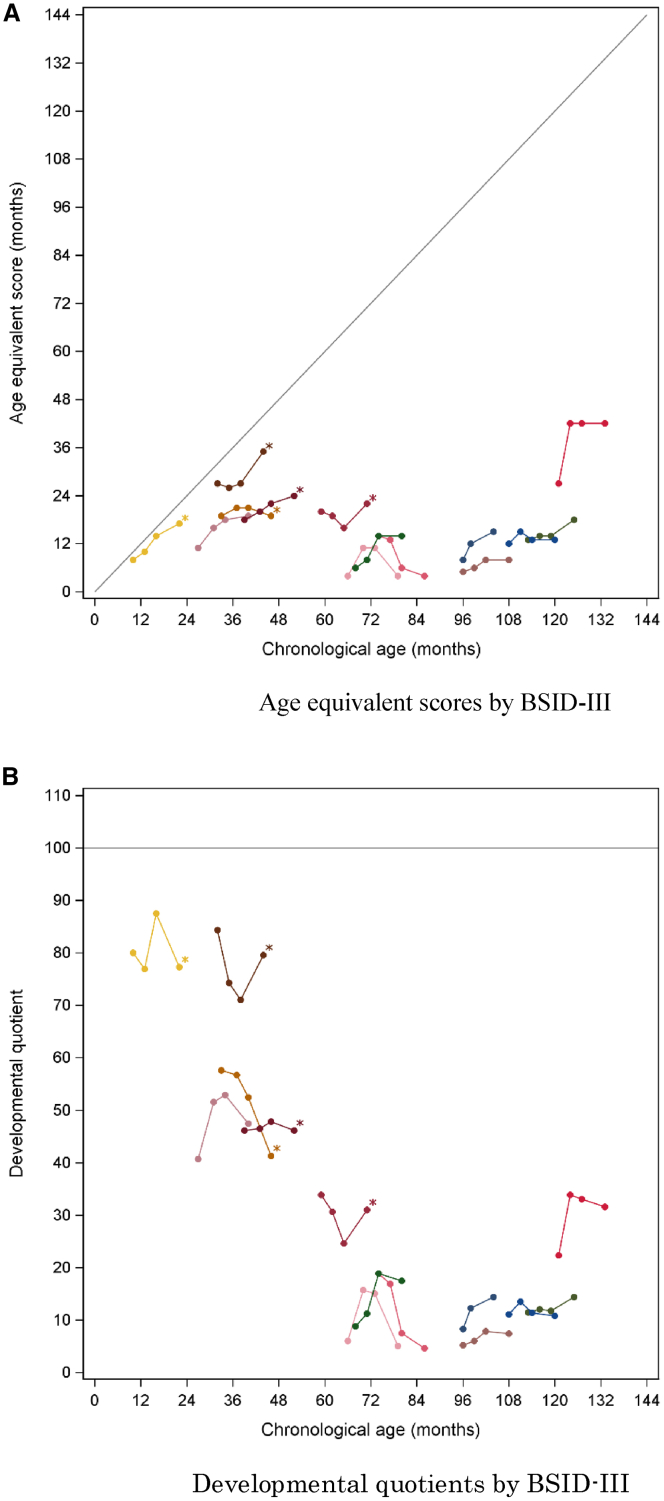
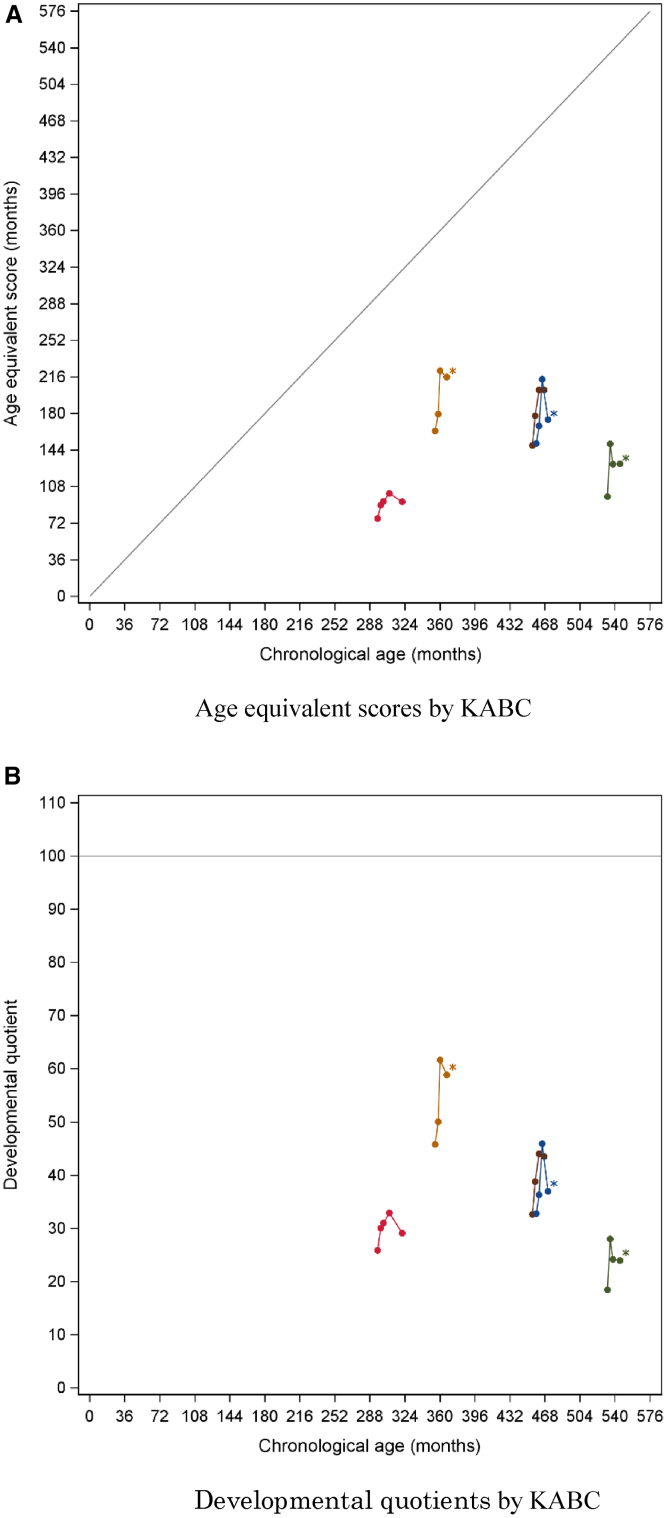
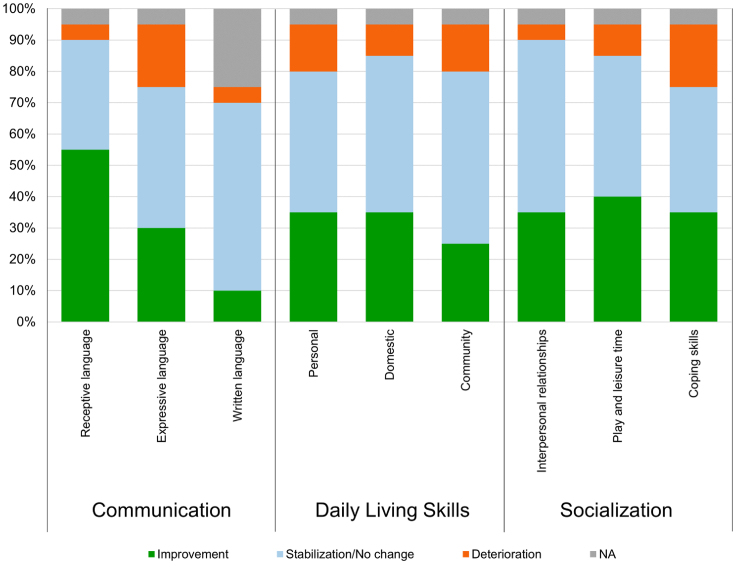
In Hunter syndrome (mucopolysaccharidosis II [MPS-II]), systemic accumulation of glycosaminoglycans (GAGs) due to a deficiency of iduronate-2-sulfatase (IDS), caused by mutations in the IDS gene, leads to multiple somatic manifestations and in patients with the severe (neuronopathic) phenotype, also to central nervous system (CNS) involvement. These symptoms cannot be effectively treated with current enzyme-replacement therapies, as they are unable to cross the blood-brain barrier (BBB). Pabinafusp alfa, a novel IDS fused with an anti-human transferrin receptor antibody, was shown to penetrate the BBB and to address neurodegeneration in preclinical studies. Subsequent phase 1/2 and 2/3 clinical studies in Japan have shown marked reduction of GAG accumulation in the cerebrospinal fluid (CSF), along with favorable clinical responses. A 26-week, open-label, randomized, parallel-group phase 2 study was conducted in Brazil to further evaluate the safety and efficacy of intravenously administered pabinafusp alfa at 1.0, 2.0, and 4.0 mg/kg/week in MPS-II patients. The safety profiles in the three dosage groups were similar. Neurodevelopmental evaluation suggested positive neurocognitive signals despite a relatively short study period. The 2.0-mg/kg group, which demonstrated marked reductions in substrate concentrations in the CSF, serum, and urine, was considered to provide the best combination regarding safety and efficacy signals.
Keywords: Hunter syndrome; JR-141; anti-human transferrin receptor antibody; blood-brain barrier; enzyme-replacement therapy; heparan sulfate; iduronate-2-sulfatase; mucopolysaccharidosis II; neurocognitive impairment; pabinafusp alfa.
Copyright © 2021 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests R.G. has been an investigator, consultant, and/or speaker within the last 12 months for Abeona, Allevex, Amicus, BioMarin, Chiesi, Denali, Idorsia, Inventiva, JCR, Lysogene, Novartis, PassageBio, PTC, RegenxBio, Sanofi-Genzyme, Sigilon, Sobi, Takeda, and Ultragenyx. A.M.M. has received honors and support for travels and congresses from BioMarin, Sanofi Genzyme, Takeda, and Ultragenyx. A.M.M. has received research fundings from Alexion, BioMarin, Sanofi Genzyme, and Takeda.
Figures
References
-
- Giuliani R. The mucopolysaccharidoses. In: Mehta A., Winchester B., editors. Lysosomal Storage Disorders: A Practical Guide. First Edition. Wiley-Blackwell; 2012. pp. 94–100.
-
- Giugliani R., Vairo F., Kubaski F., Poswar F., Riegel M., Baldo G., Saute J.A. Neurological manifestations of lysosomal disorders and emerging therapies targeting the CNS. Lancet Child Adolesc. Health. 2018;2:56–68. - PubMed
-
- Muenzer J., Wraith J.E., Beck M., Giugliani R., Harmatz P., Eng C.M., Vellodi A., Martin R., Ramaswami U., Gucsavas-Calikoglu M. A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome) Genet. Med. 2006;8:465–473. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
