

The SARS-CoV-2 nucleocapsid protein is dynamic, disordered, and phase separates with RNA
- PMID: 33782395
- PMCID: PMC8007728
- DOI: 10.1038/s41467-021-21953-3
The SARS-CoV-2 nucleocapsid protein is dynamic, disordered, and phase separates with RNA
Abstract
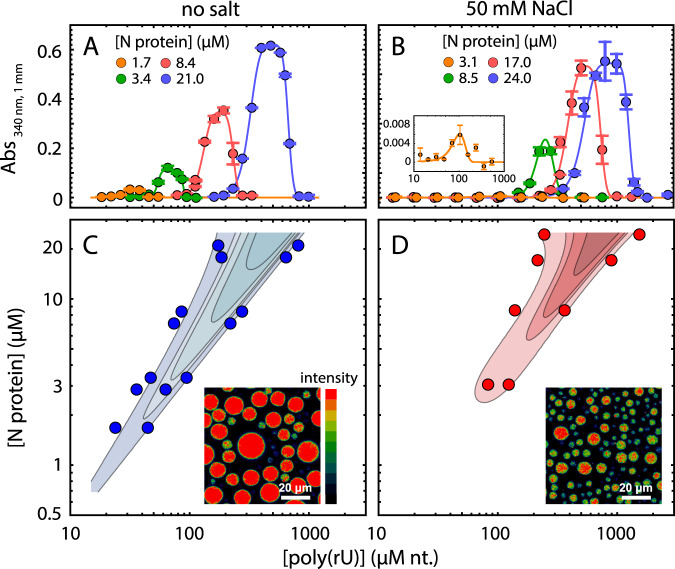
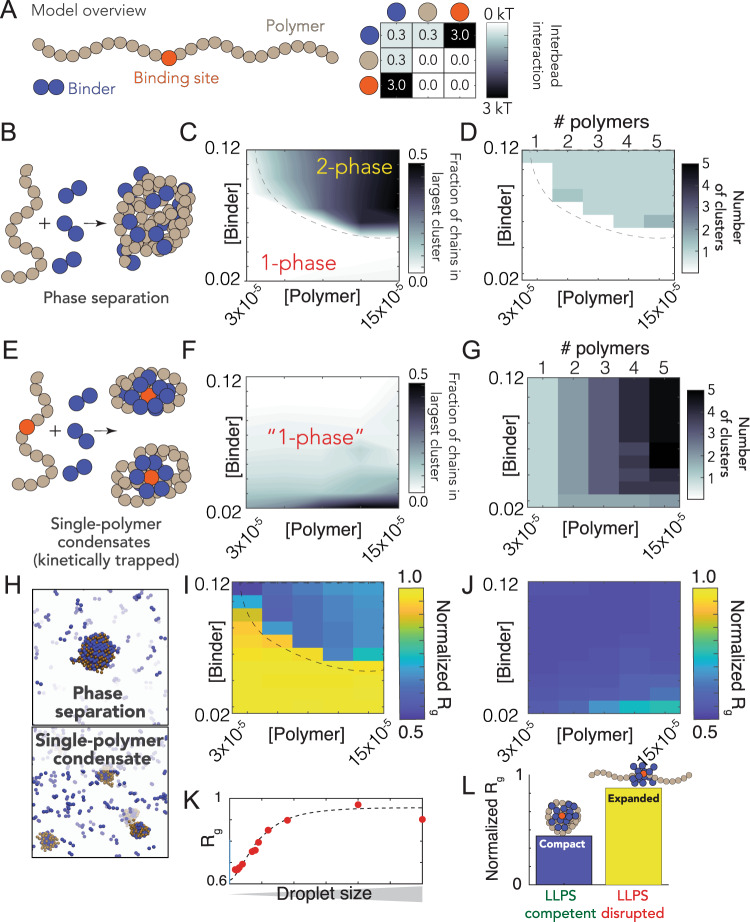
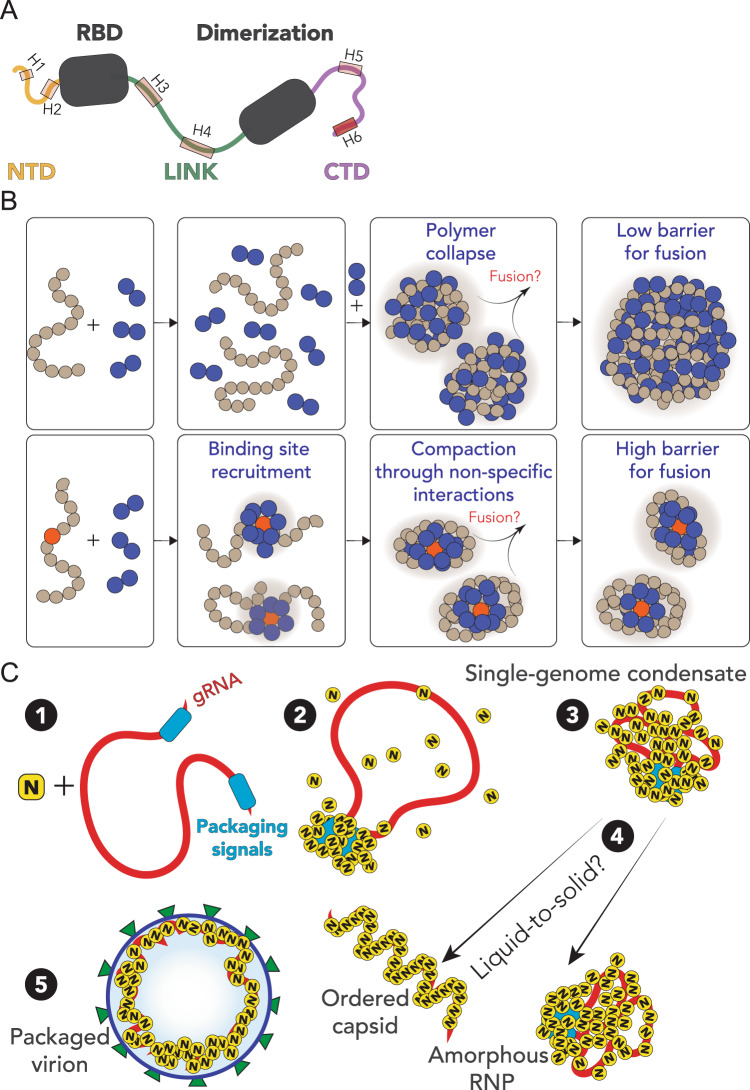
The SARS-CoV-2 nucleocapsid (N) protein is an abundant RNA-binding protein critical for viral genome packaging, yet the molecular details that underlie this process are poorly understood. Here we combine single-molecule spectroscopy with all-atom simulations to uncover the molecular details that contribute to N protein function. N protein contains three dynamic disordered regions that house putative transiently-helical binding motifs. The two folded domains interact minimally such that full-length N protein is a flexible and multivalent RNA-binding protein. N protein also undergoes liquid-liquid phase separation when mixed with RNA, and polymer theory predicts that the same multivalent interactions that drive phase separation also engender RNA compaction. We offer a simple symmetry-breaking model that provides a plausible route through which single-genome condensation preferentially occurs over phase separation, suggesting that phase separation offers a convenient macroscopic readout of a key nanoscopic interaction.
Conflict of interest statement
A.S.H. is a scientific consultant with Dewpoint Therapeutics. This affiliation in no way influenced the content of this study. All other authors declare no competing interests.
Figures
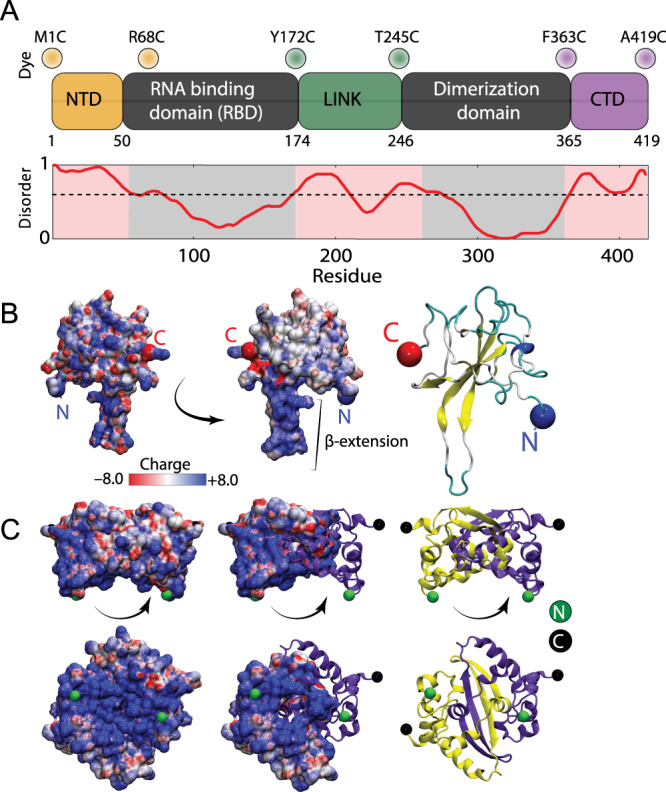
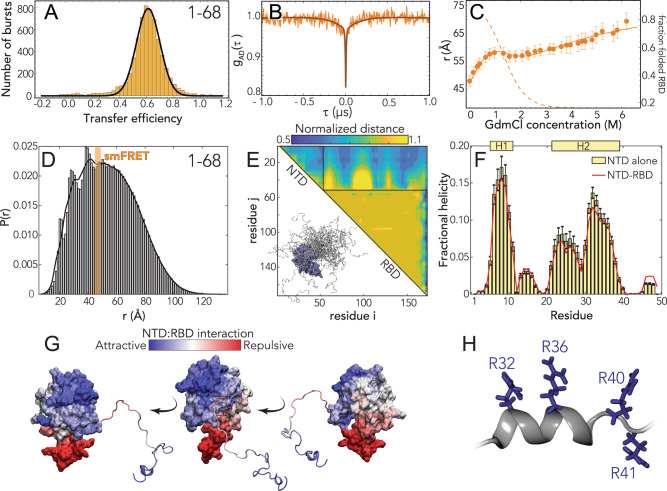
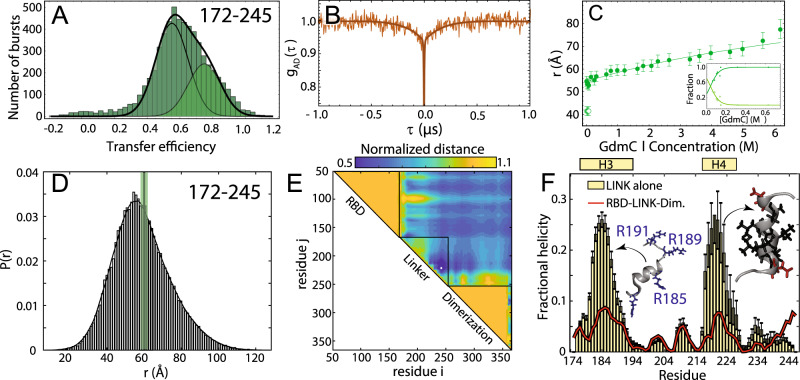
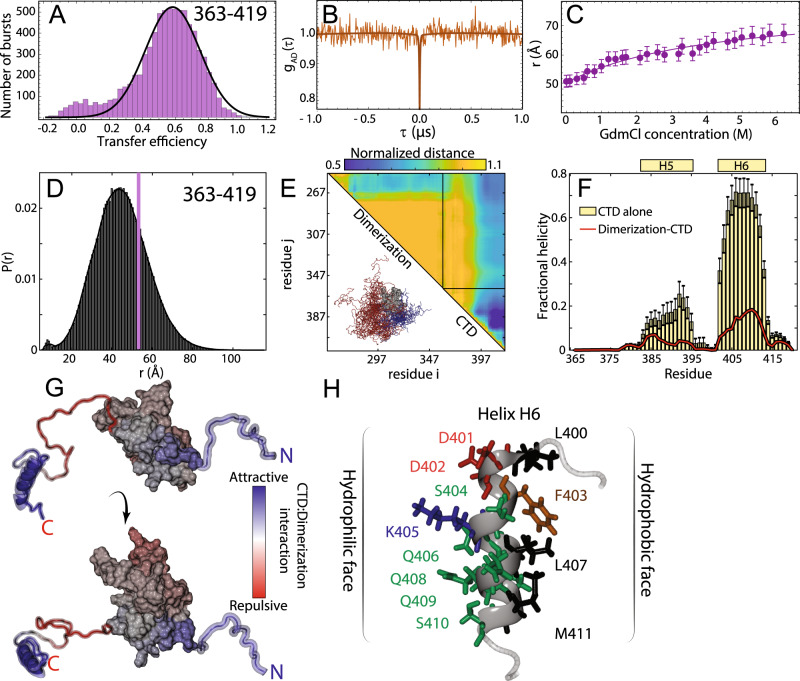
Update of
-
The SARS-CoV-2 nucleocapsid protein is dynamic, disordered, and phase separates with RNA.bioRxiv [Preprint]. 2020 Dec 21:2020.06.17.158121. doi: 10.1101/2020.06.17.158121. bioRxiv. 2020. Update in: Nat Commun. 2021 Mar 29;12(1):1936. doi: 10.1038/s41467-021-21953-3. PMID: 32587966 Free PMC article. Updated. Preprint.
References
-
- Roser, M., Ritchie, H., Ortiz-Ospina, E. & Hasell, J. Coronavirus Pandemic (COVID-19). Our World in Data (2020).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
