

A unified haplotype-based method for accurate and comprehensive variant calling
- PMID: 33782612
- PMCID: PMC7611855
- DOI: 10.1038/s41587-021-00861-3
A unified haplotype-based method for accurate and comprehensive variant calling
Abstract
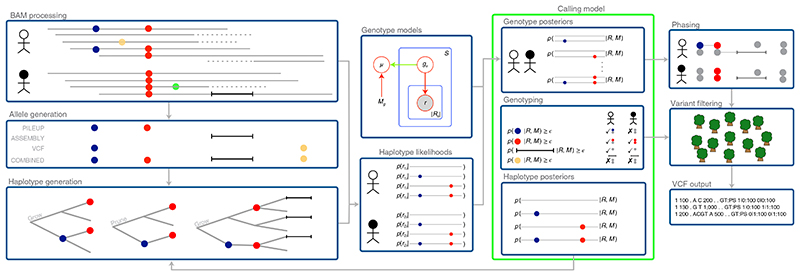
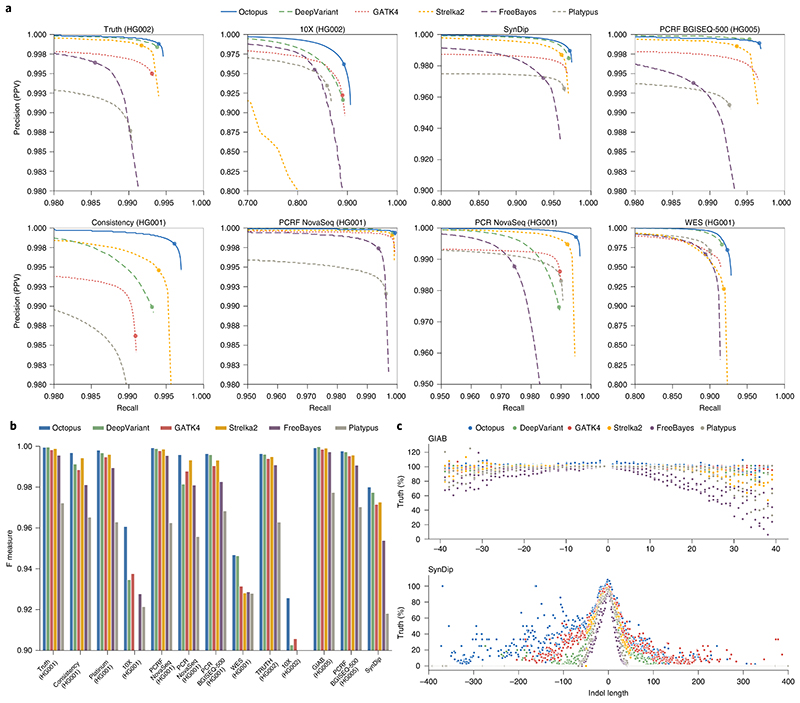
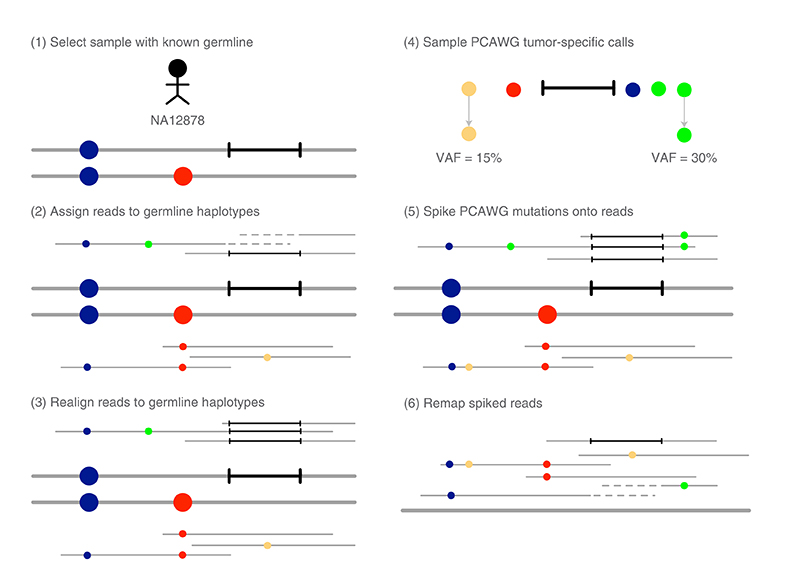
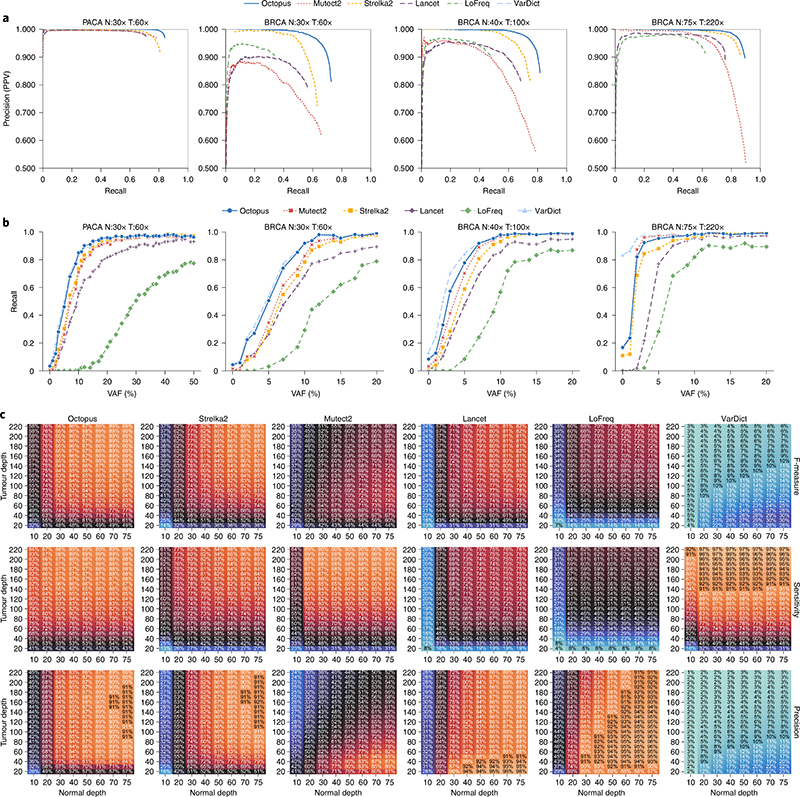
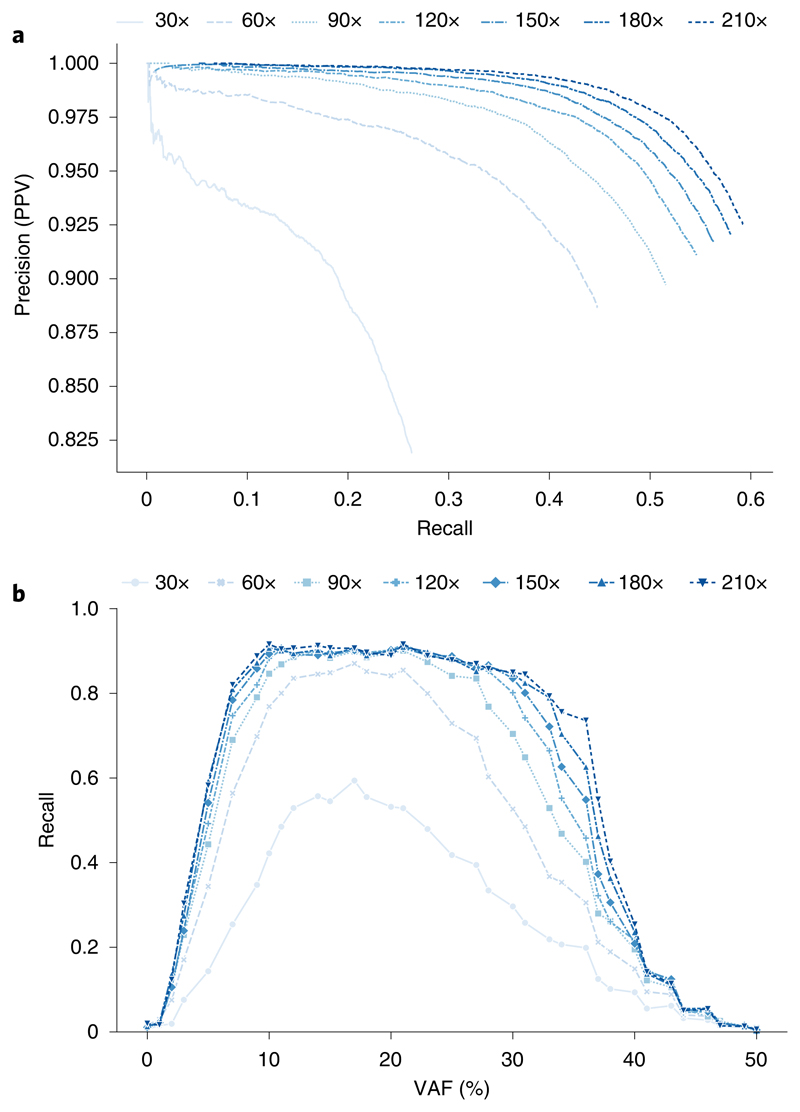
Almost all haplotype-based variant callers were designed specifically for detecting common germline variation in diploid populations, and give suboptimal results in other scenarios. Here we present Octopus, a variant caller that uses a polymorphic Bayesian genotyping model capable of modeling sequencing data from a range of experimental designs within a unified haplotype-aware framework. Octopus combines sequencing reads and prior information to phase-called genotypes of arbitrary ploidy, including those with somatic mutations. We show that Octopus accurately calls germline variants in individuals, including single nucleotide variants, indels and small complex replacements such as microinversions. Using a synthetic tumor data set derived from clean sequencing data from a sample with known germline haplotypes and observed mutations in a large cohort of tumor samples, we show that Octopus is more sensitive to low-frequency somatic variation, yet calls considerably fewer false positives than other methods. Octopus also outputs realigned evidence BAM files to aid validation and interpretation.
© 2021. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Kim S, et al. Strelka2: fast and accurate calling of germline and somatic variants. Nat Methods. 2018;15:591–594. - PubMed
-
- Poplin R, et al. A universal SNP and small-indel variant caller using deep neural networks. Nat Biotechnol. 2018;36:983–987. - PubMed
-
- Garrison E, Marth G. Haplotype-based variant detection from short-read sequencing. 2012 Preprint at https://arxiv.org/abs/1207.3907.
Publication types
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
