

Interaction of Aromatic Molecules with Forsterite: Accuracy of the Periodic DFT-D4 Method
- PMID: 33784098
- PMCID: PMC8154625
- DOI: 10.1021/acs.jpca.1c02326
Interaction of Aromatic Molecules with Forsterite: Accuracy of the Periodic DFT-D4 Method
Abstract
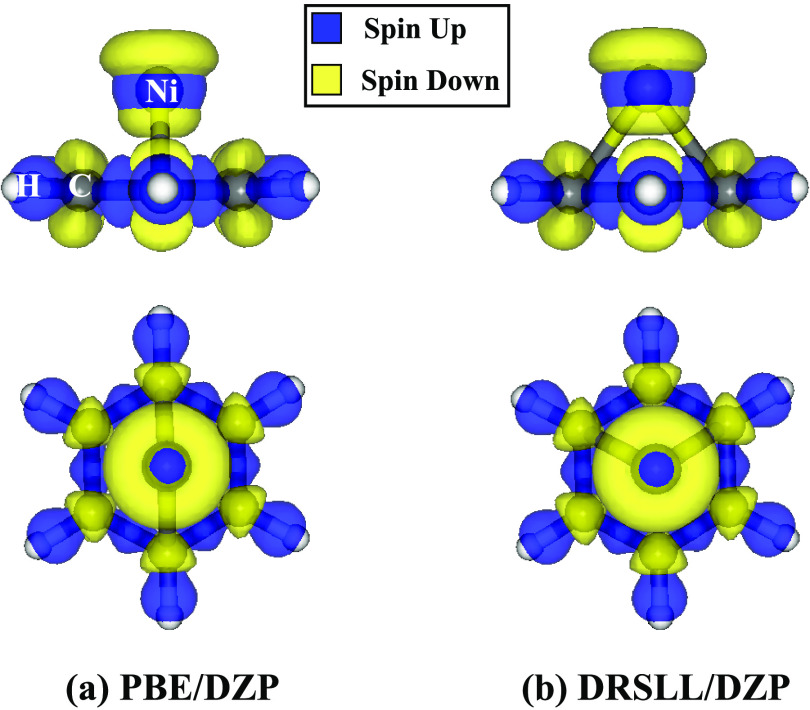
Density functional theory (DFT) has provided deep atomic-level insights into the adsorption behavior of aromatic molecules on solid surfaces. However, modeling the surface phenomena of large molecules on mineral surfaces with accurate plane wave methods (PW) can be orders of magnitude more computationally expensive than localized atomic orbitals (LCAO) methods. In the present work, we propose a less costly approach based on the DFT-D4 method (PBE-D4), using LCAO, to study the interactions of aromatic molecules with the {010} forsterite (Mg2SiO4) surface for their relevance in astrochemistry. We studied the interaction of benzene with the pristine {010} forsterite surface and with transition-metal cations (Fe2+ and Ni2+) using PBE-D4 and a vdW-inclusive density functional (Dion, Rydberg, Schröder, Langreth, and Lundqvist (DRSLL)) with LCAO methods. PBE-D4 shows good agreement with coupled-cluster methods (CCSD(T)) for the binding energy trend of cation complexes and with PW methods for the binding energy of benzene on the forsterite surface with a difference of about 0.03 eV. The basis set superposition error (BSSE) correction is shown to be essential to ensure a correct estimation of the binding energies even when large basis sets are employed for single-point calculations of the optimized structures with smaller basis sets. We also studied the interaction of naphthalene and benzocoronene on pristine and transition-metal-doped {010} forsterite surfaces as a test case for PBE-D4. Yielding results that are in good agreement with the plane wave methods with a difference of about 0.02-0.17 eV, the PBE-D4 method is demonstrated to be effective in unraveling the binding structures and the energetic trends of aromatic molecules on pristine and transition-metal-doped forsterite mineral surfaces. Furthermore, PBE-D4 results are in good agreement with its predecessor PBE-D3(BJM) and with the vdW-inclusive density functionals, as long as transition metals are not involved. Hence, PBE-D4/CP-DZP has been proven to be a robust theory level to study the interaction of aromatic molecules on mineral surfaces.
Conflict of interest statement
The authors declare no competing financial interest.
Figures








References
-
- Tielens A. G. G. M. The Molecular Universe. Rev. Mod. Phys. 2013, 85, 1021–1081. 10.1103/RevModPhys.85.1021. - DOI
-
- Sephton M.Treatise on Geochemistry (Second Edition), second ed.; Holland H. D.; Turekian K. K., Eds.; Elsevier: Oxford, 2014; pp 1–31.
-
- Tielens A. G. G. M. Interstellar Polycyclic Aromatic Hydrocarbon Molecules. Annu. Rev. Astron. Astrophys. 2008, 46, 289–337. 10.1146/annurev.astro.46.060407.145211. - DOI
-
- Rauls E.; Hornekær L. Catalyzed Routes to Molecular Hydrogen Formation and Hydrogen Addition Reactions on Neutral Polycyclic Aromatic Hydrocarbons under Interstellar Conditions. Astrophys. J. 2008, 679, 531–536. 10.1086/587614. - DOI
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
