

KDM5 inhibition offers a novel therapeutic strategy for the treatment of KMT2D mutant lymphomas
- PMID: 33786580
- PMCID: PMC8351530
- DOI: 10.1182/blood.2020008743
KDM5 inhibition offers a novel therapeutic strategy for the treatment of KMT2D mutant lymphomas
Erratum in
-
Heward J, Konali L, D'Avola A, et al. KDM5 inhibition offers a novel therapeutic strategy for the treatment of KMT2D mutant lymphomas. Blood. 2021;138(5):370-381.Blood. 2022 Sep 8;140(10):1183. doi: 10.1182/blood.2022016495. Blood. 2022. PMID: 36074529 Free PMC article. No abstract available.
Abstract
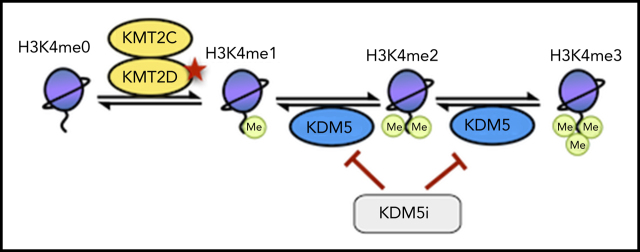
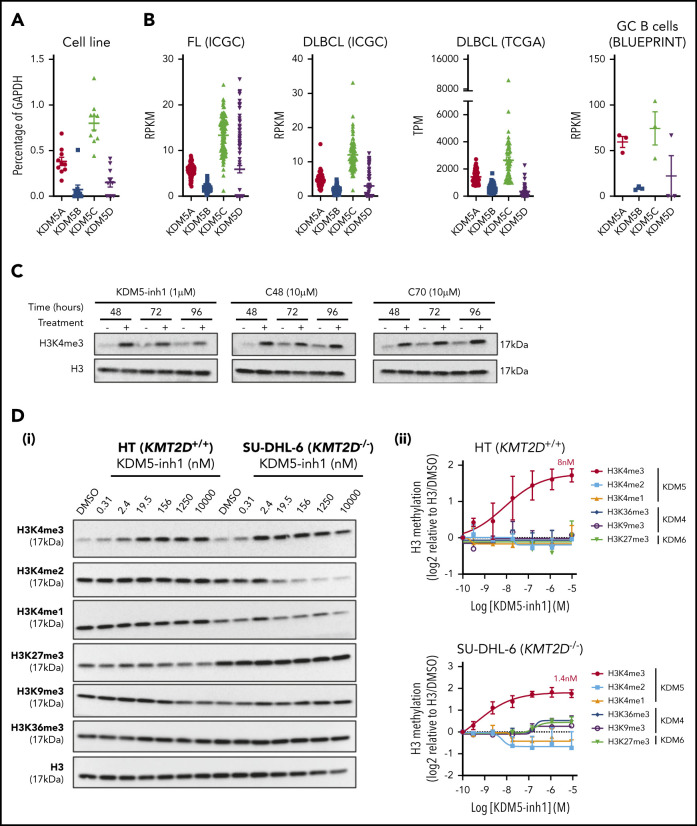
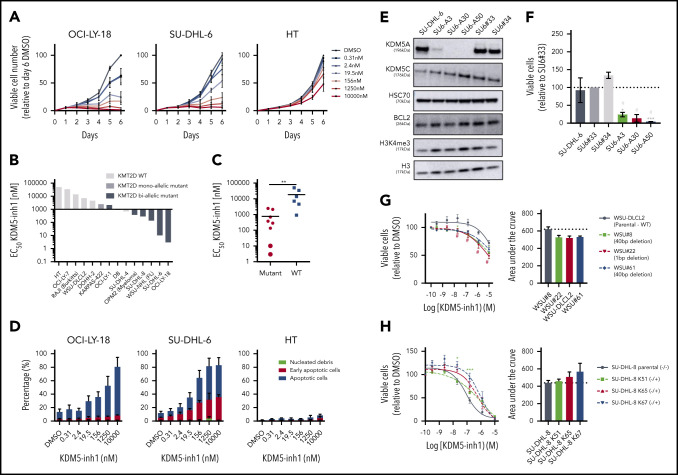
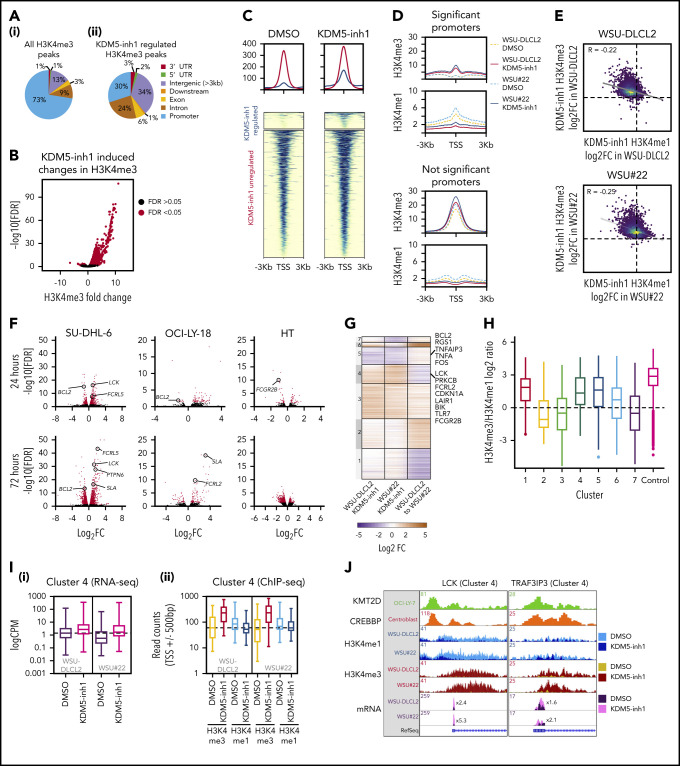
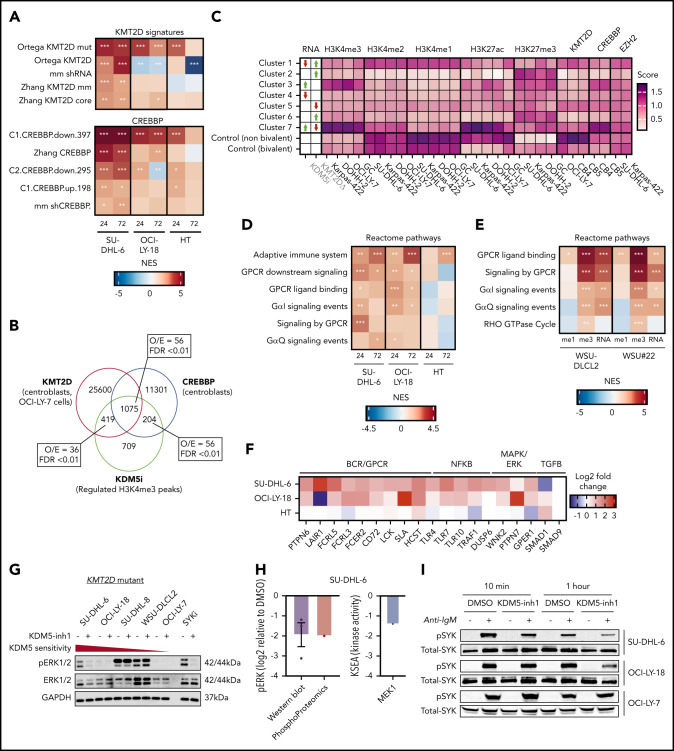
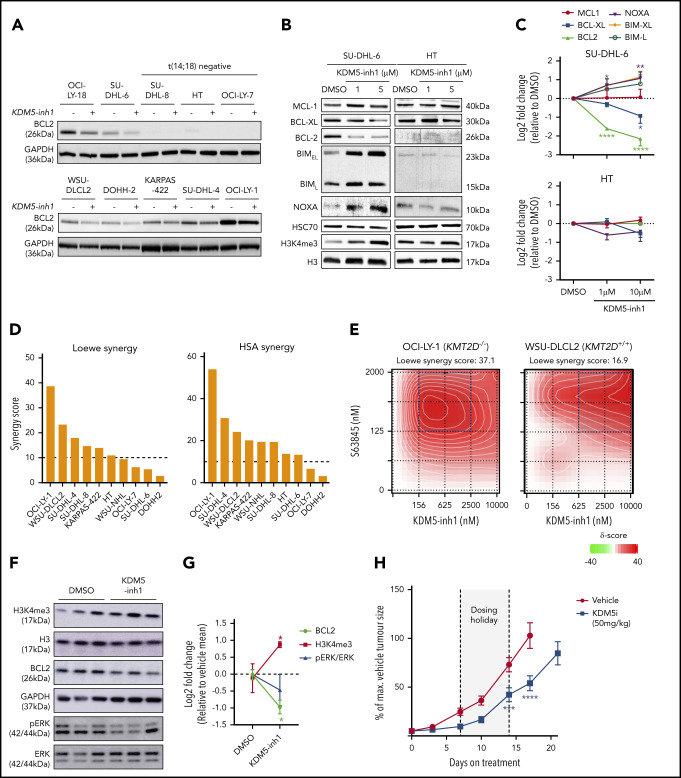
Loss-of-function mutations in KMT2D are a striking feature of germinal center (GC) lymphomas, resulting in decreased histone 3 lysine 4 (H3K4) methylation and altered gene expression. We hypothesized that inhibition of the KDM5 family, which demethylates H3K4me3/me2, would reestablish H3K4 methylation and restore the expression of genes repressed on loss of KMT2D. KDM5 inhibition increased H3K4me3 levels and caused an antiproliferative response in vitro, which was markedly greater in both endogenous and gene-edited KMT2D mutant diffuse large B-cell lymphoma cell lines, whereas tumor growth was inhibited in KMT2D mutant xenografts in vivo. KDM5 inhibition reactivated both KMT2D-dependent and -independent genes, resulting in diminished B-cell signaling and altered expression of B-cell lymphoma 2 (BCL2) family members, including BCL2 itself. KDM5 inhibition may offer an effective therapeutic strategy for ameliorating KMT2D loss-of-function mutations in GC lymphomas.
© 2021 by The American Society of Hematology.
Figures
Comment in
-
Epigenetic balance in DLBCL.Blood. 2021 Aug 5;138(5):355-356. doi: 10.1182/blood.2021011647. Blood. 2021. PMID: 34351369 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
