

Loss of the abasic site sensor HMCES is synthetic lethal with the activity of the APOBEC3A cytosine deaminase in cancer cells
- PMID: 33788831
- PMCID: PMC8041192
- DOI: 10.1371/journal.pbio.3001176
Loss of the abasic site sensor HMCES is synthetic lethal with the activity of the APOBEC3A cytosine deaminase in cancer cells
Abstract
Analysis of cancer mutagenic signatures provides information about the origin of mutations and can inform the use of clinical therapies, including immunotherapy. In particular, APOBEC3A (A3A) has emerged as a major driver of mutagenesis in cancer cells, and its expression results in DNA damage and susceptibility to treatment with inhibitors of the ATR and CHK1 checkpoint kinases. Here, we report the implementation of CRISPR/Cas-9 genetic screening to identify susceptibilities of multiple A3A-expressing lung adenocarcinoma (LUAD) cell lines. We identify HMCES, a protein recently linked to the protection of abasic sites, as a central protein for the tolerance of A3A expression. HMCES depletion results in synthetic lethality with A3A expression preferentially in a TP53-mutant background. Analysis of previous screening data reveals a strong association between A3A mutational signatures and sensitivity to HMCES loss and indicates that HMCES is specialized in protecting against a narrow spectrum of DNA damaging agents in addition to A3A. We experimentally show that both HMCES disruption and A3A expression increase susceptibility of cancer cells to ionizing radiation (IR), oxidative stress, and ATR inhibition, strategies that are often applied in tumor therapies. Overall, our results suggest that HMCES is an attractive target for selective treatment of A3A-expressing tumors.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
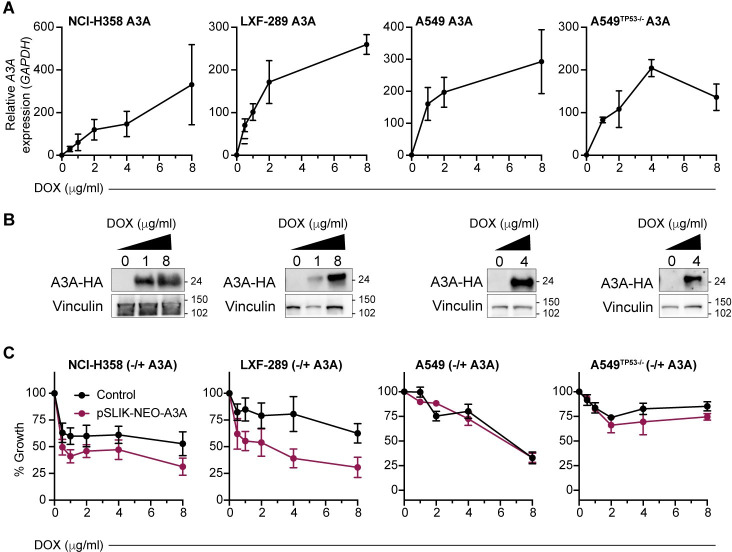
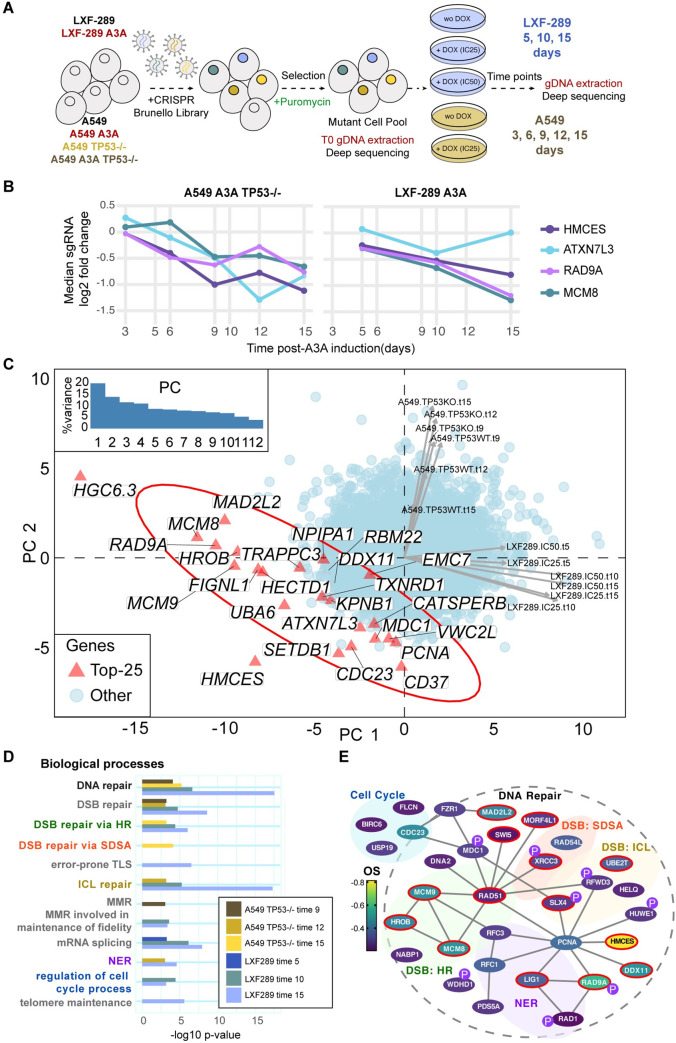
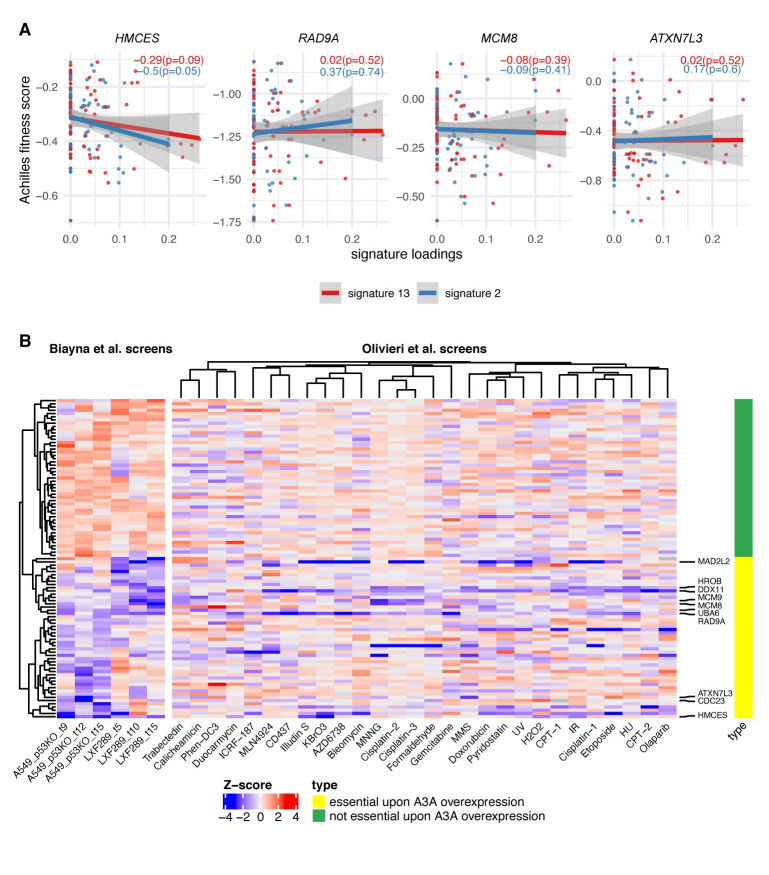
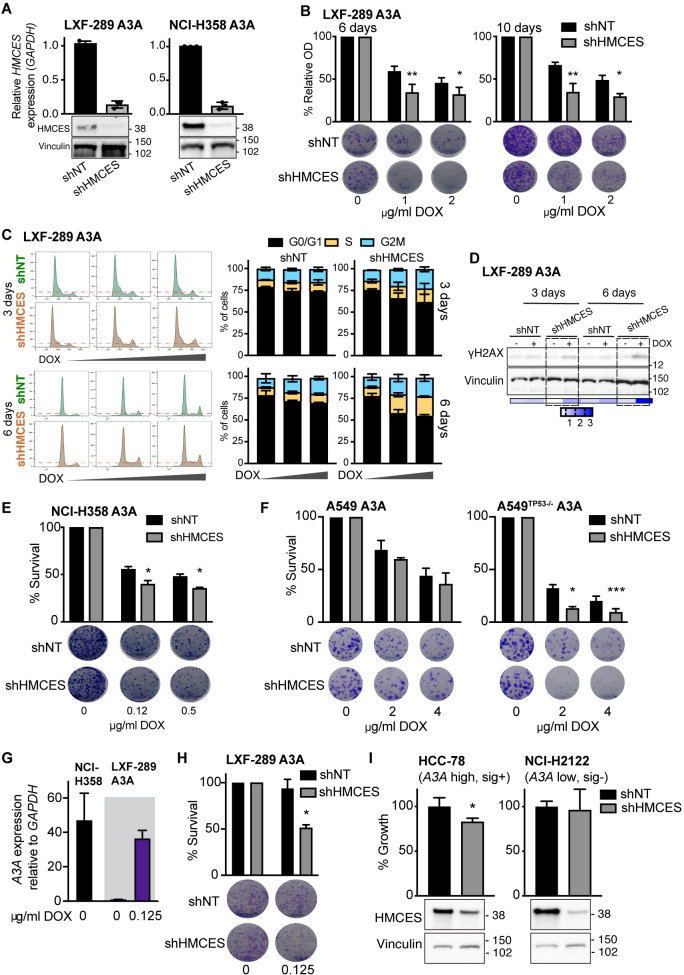
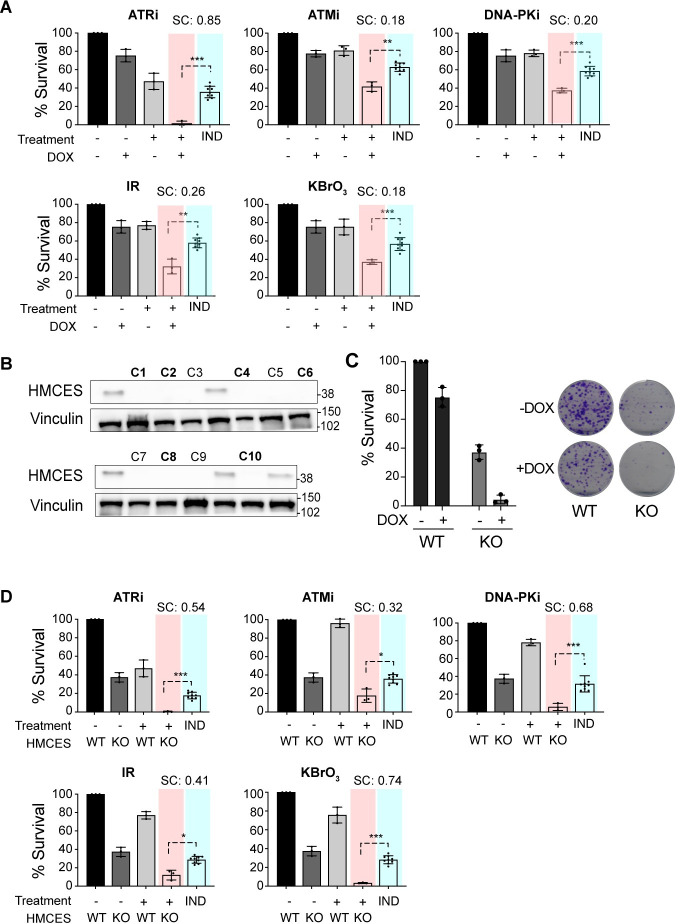
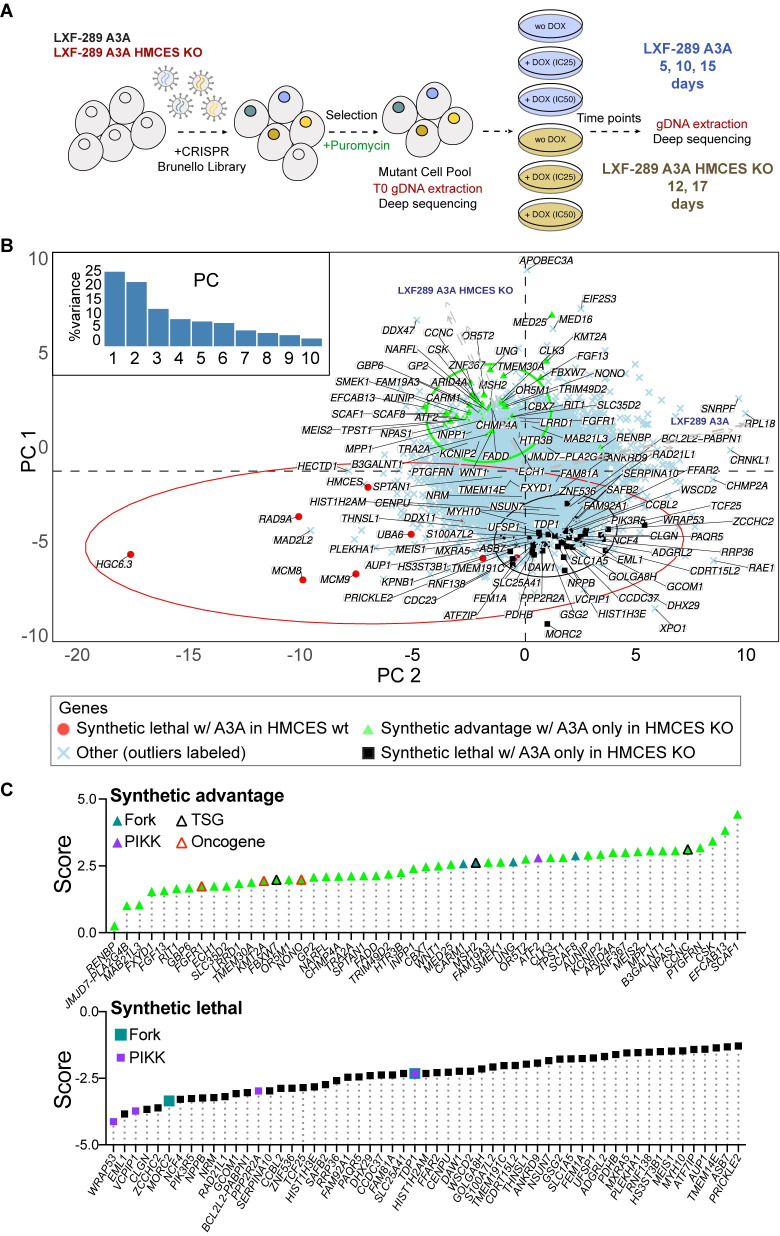
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
