

Population genomics of the pathogenic yeast Candida tropicalis identifies hybrid isolates in environmental samples
- PMID: 33788904
- PMCID: PMC8041210
- DOI: 10.1371/journal.ppat.1009138
Population genomics of the pathogenic yeast Candida tropicalis identifies hybrid isolates in environmental samples
Abstract
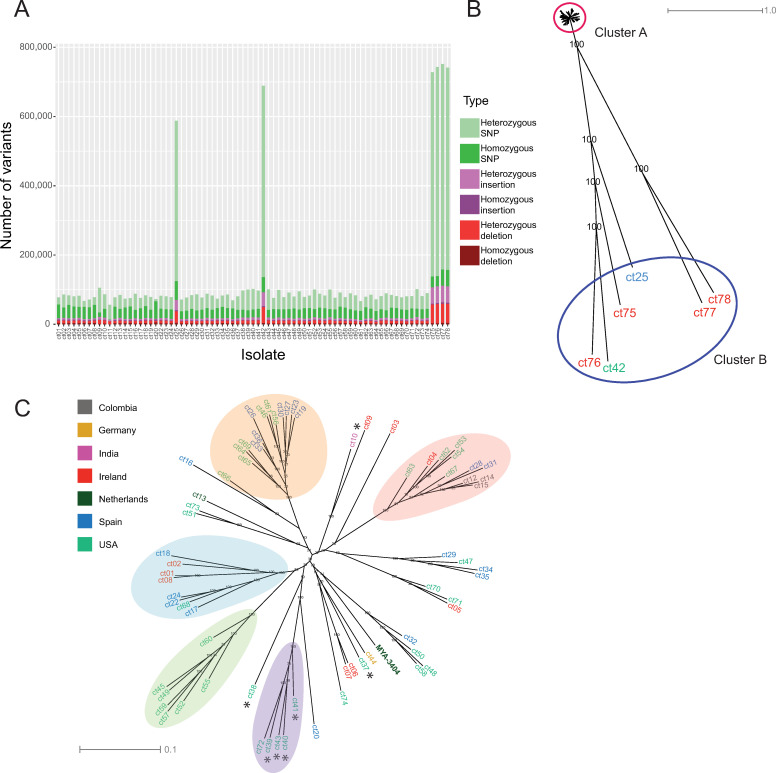
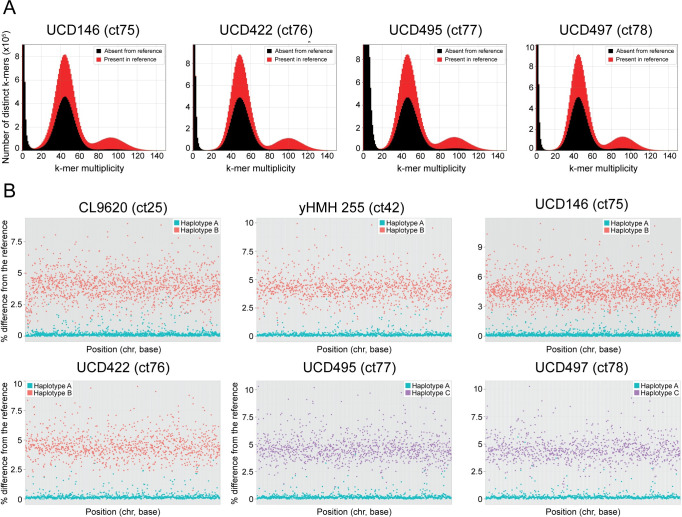
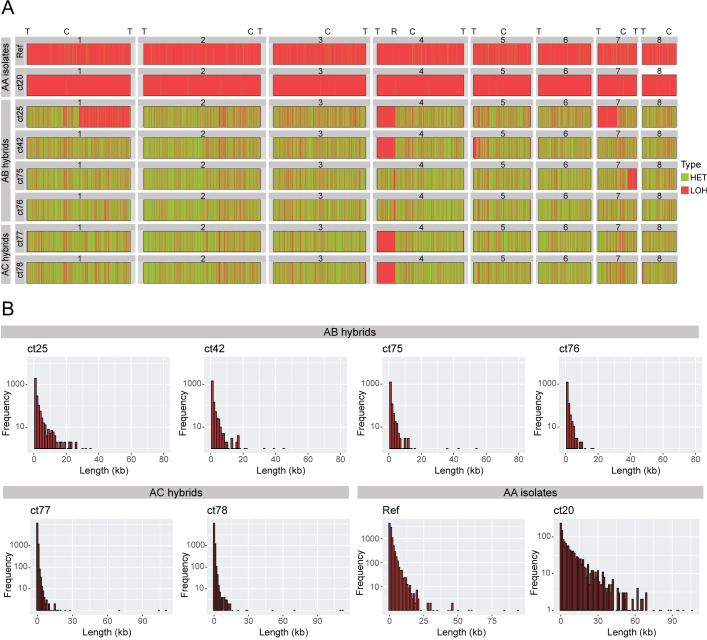
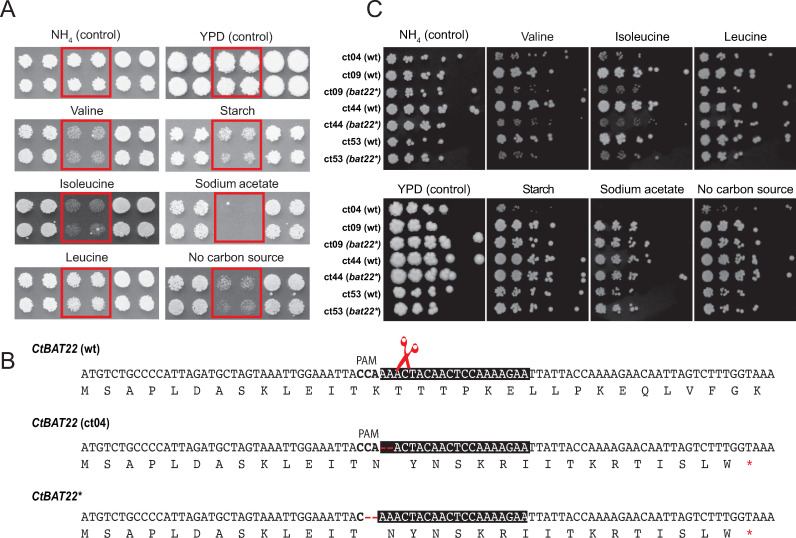
Candida tropicalis is a human pathogen that primarily infects the immunocompromised. Whereas the genome of one isolate, C. tropicalis MYA-3404, was originally sequenced in 2009, there have been no large-scale, multi-isolate studies of the genetic and phenotypic diversity of this species. Here, we used whole genome sequencing and phenotyping to characterize 77 isolates of C. tropicalis from clinical and environmental sources from a variety of locations. We show that most C. tropicalis isolates are diploids with approximately 2-6 heterozygous variants per kilobase. The genomes are relatively stable, with few aneuploidies. However, we identified one highly homozygous isolate and six isolates of C. tropicalis with much higher heterozygosity levels ranging from 36-49 heterozygous variants per kilobase. Our analyses show that the heterozygous isolates represent two different hybrid lineages, where the hybrids share one parent (A) with most other C. tropicalis isolates, but the second parent (B or C) differs by at least 4% at the genome level. Four of the sequenced isolates descend from an AB hybridization, and two from an AC hybridization. The hybrids are MTLa/α heterozygotes. Hybridization, or mating, between different parents is therefore common in the evolutionary history of C. tropicalis. The new hybrids were predominantly found in environmental niches, including from soil. Hybridization is therefore unlikely to be associated with virulence. In addition, we used genotype-phenotype correlation and CRISPR-Cas9 editing to identify a genome variant that results in the inability of one isolate to utilize certain branched-chain amino acids as a sole nitrogen source.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Candida tropicalis is the most prevalent yeast species causing candidemia in Algeria: the urgent need for antifungal stewardship and infection control measures.Antimicrob Resist Infect Control. 2020 Apr 7;9(1):50. doi: 10.1186/s13756-020-00710-z. Antimicrob Resist Infect Control. 2020. PMID: 32264966 Free PMC article.
-
Phenotypic switching in Candida tropicalis: association with modification of putative virulence attributes and antifungal drug sensitivity.Med Mycol. 2014 Jan;52(1):106-14. doi: 10.3109/13693786.2013.825822. Med Mycol. 2014. PMID: 23971864
-
Analysis of global antifungal surveillance results reveals predominance of Erg11 Y132F alteration among azole-resistant Candida parapsilosis and Candida tropicalis and country-specific isolate dissemination.Int J Antimicrob Agents. 2020 Jan;55(1):105799. doi: 10.1016/j.ijantimicag.2019.09.003. Epub 2019 Sep 11. Int J Antimicrob Agents. 2020. PMID: 31520783
-
The yeast, the antifungal, and the wardrobe: a journey into antifungal resistance mechanisms of Candida tropicalis.Can J Microbiol. 2020 Jun;66(6):377-388. doi: 10.1139/cjm-2019-0531. Epub 2020 Apr 22. Can J Microbiol. 2020. PMID: 32319304 Review.
-
Insights into Candida tropicalis nosocomial infections and virulence factors.Eur J Clin Microbiol Infect Dis. 2012 Jul;31(7):1399-412. doi: 10.1007/s10096-011-1455-z. Epub 2011 Oct 30. Eur J Clin Microbiol Infect Dis. 2012. PMID: 22037823 Review.
Cited by
-
Biology and genetic diversity of Candida krusei isolates from fermented vegetables and clinical samples in China.Virulence. 2024 Dec;15(1):2411543. doi: 10.1080/21505594.2024.2411543. Epub 2024 Oct 17. Virulence. 2024. PMID: 39359062 Free PMC article.
-
Multilocus sequence typing (MLST) analysis reveals many novel genotypes and a high level of genetic diversity in Candida tropicalis isolates from Italy and Africa.Mycoses. 2022 Nov;65(11):989-1000. doi: 10.1111/myc.13483. Epub 2022 Jul 7. Mycoses. 2022. PMID: 35713604 Free PMC article.
-
Variations in candidalysin amino acid sequence influence toxicity and host responses.mBio. 2024 Aug 14;15(8):e0335123. doi: 10.1128/mbio.03351-23. Epub 2024 Jul 2. mBio. 2024. PMID: 38953356 Free PMC article.
-
Clonality, inbreeding, and hybridization in two extremotolerant black yeasts.Gigascience. 2022 Oct 6;11:giac095. doi: 10.1093/gigascience/giac095. Gigascience. 2022. PMID: 36200832 Free PMC article.
-
Whole Genome Sequencing Shows Genetic Diversity, as Well as Clonal Complex and Gene Polymorphisms Associated with Fluconazole Non-Susceptible Isolates of Candida tropicalis.J Fungi (Basel). 2022 Aug 23;8(9):896. doi: 10.3390/jof8090896. J Fungi (Basel). 2022. PMID: 36135621 Free PMC article.
References
-
- Pfaller MA, Diekema DJ, Gibbs DL, Newell VA, Ellis D, Tullio V, et al.. Results from the ARTEMIS DISK Global Antifungal Surveillance Study, 1997 to 2007: a 10.5-year analysis of susceptibilities of Candida Species to fluconazole and voriconazole as determined by CLSI standardized disk diffusion. J Clin Microbiol. 2010;48: 1366–1377. 10.1128/JCM.02117-09 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
