

Estimation of Secondary Household Attack Rates for Emergent Spike L452R Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Variants Detected by Genomic Surveillance at a Community-Based Testing Site in San Francisco
- PMID: 33788923
- PMCID: PMC8083548
- DOI: 10.1093/cid/ciab283
Estimation of Secondary Household Attack Rates for Emergent Spike L452R Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Variants Detected by Genomic Surveillance at a Community-Based Testing Site in San Francisco
Abstract
Background: Sequencing of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) viral genome from patient samples is an important epidemiological tool for monitoring and responding to the pandemic, including the emergence of new mutations in specific communities.
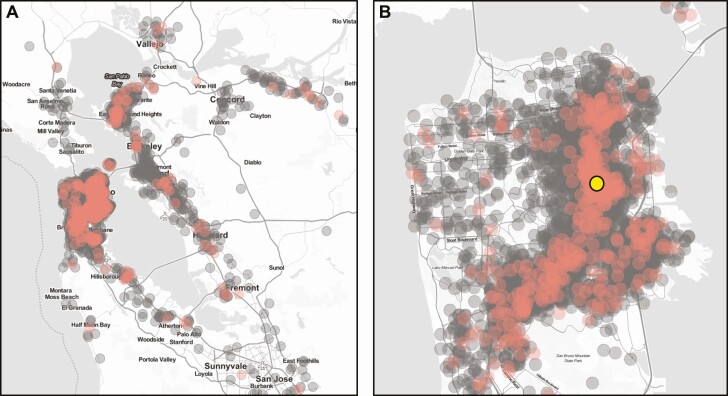
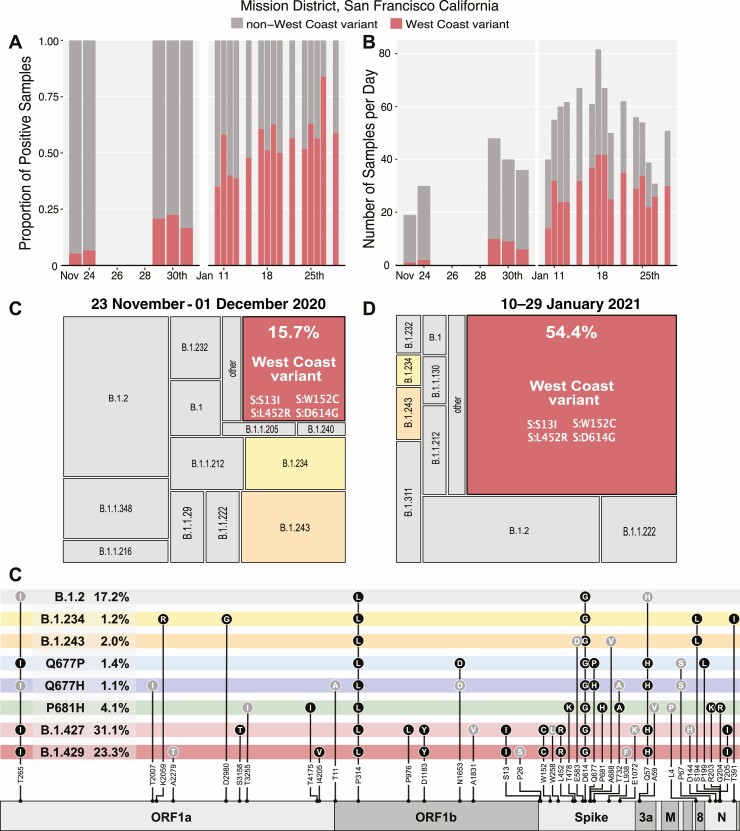
Methods: SARS-CoV-2 genomic sequences were generated from positive samples collected, along with epidemiological metadata, at a walk-up, rapid testing site in the Mission District of San Francisco, California during 22 November to 1 December, 2020, and 10-29 January 2021. Secondary household attack rates and mean sample viral load were estimated and compared across observed variants.
Results: A total of 12 124 tests were performed yielding 1099 positives. From these, 928 high-quality genomes were generated. Certain viral lineages bearing spike mutations, defined in part by L452R, S13I, and W152C, comprised 54.4% of the total sequences from January, compared to 15.7% in November. Household contacts exposed to the "California" or "West Coast" variants (B.1.427 and B.1.429) were at higher risk of infection compared to household contacts exposed to lineages lacking these variants (0.36 vs 0.29, risk ratio [RR] = 1.28; 95% confidence interval [CI]: 1.00-1.64). The reproductive number was estimated to be modestly higher than other lineages spreading in California during the second half of 2020. Viral loads were similar among persons infected with West Coast versus non-West Coast strains, as was the proportion of individuals with symptoms (60.9% vs 64.3%).
Conclusions: The increase in prevalence, relative household attack rates, and reproductive number are consistent with a modest transmissibility increase of the West Coast variants. Summary: We observed a growing prevalence and modestly elevated attack rate for "West Coast" severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) variants in a community testing setting in San Francisco during January 2021, suggesting its modestly higher transmissibility.
Keywords: SARS-CoV-2; household transmission; secondary attack rates; spike mutation; variant.
© The Author(s) 2021. Published by Oxford University Press for the Infectious Diseases Society of America.
Figures
Update of
-
Estimation of secondary household attack rates for emergent SARS-CoV-2 variants detected by genomic surveillance at a community-based testing site in San Francisco.medRxiv [Preprint]. 2021 Mar 3:2021.03.01.21252705. doi: 10.1101/2021.03.01.21252705. medRxiv. 2021. Update in: Clin Infect Dis. 2022 Jan 7;74(1):32-39. doi: 10.1093/cid/ciab283. PMID: 33688689 Free PMC article. Updated. Preprint.
Similar articles
-
Estimation of secondary household attack rates for emergent SARS-CoV-2 variants detected by genomic surveillance at a community-based testing site in San Francisco.medRxiv [Preprint]. 2021 Mar 3:2021.03.01.21252705. doi: 10.1101/2021.03.01.21252705. medRxiv. 2021. Update in: Clin Infect Dis. 2022 Jan 7;74(1):32-39. doi: 10.1093/cid/ciab283. PMID: 33688689 Free PMC article. Updated. Preprint.
-
Community Transmission of Severe Acute Respiratory Syndrome Coronavirus 2 Disproportionately Affects the Latinx Population During Shelter-in-Place in San Francisco.Clin Infect Dis. 2021 Jul 30;73(Suppl 2):S127-S135. doi: 10.1093/cid/ciaa1234. Clin Infect Dis. 2021. PMID: 32821935 Free PMC article.
-
Acquisition of the L452R Mutation in the ACE2-Binding Interface of Spike Protein Triggers Recent Massive Expansion of SARS-CoV-2 Variants.J Clin Microbiol. 2021 Oct 19;59(11):e0092121. doi: 10.1128/JCM.00921-21. Epub 2021 Aug 11. J Clin Microbiol. 2021. PMID: 34379531 Free PMC article.
-
Household Transmission of SARS-CoV-2: A Systematic Review and Meta-analysis.JAMA Netw Open. 2020 Dec 1;3(12):e2031756. doi: 10.1001/jamanetworkopen.2020.31756. JAMA Netw Open. 2020. PMID: 33315116 Free PMC article.
-
Factors Associated With Household Transmission of SARS-CoV-2: An Updated Systematic Review and Meta-analysis.JAMA Netw Open. 2021 Aug 2;4(8):e2122240. doi: 10.1001/jamanetworkopen.2021.22240. JAMA Netw Open. 2021. PMID: 34448865 Free PMC article.
Cited by
-
Online Phylogenetics with matOptimize Produces Equivalent Trees and is Dramatically More Efficient for Large SARS-CoV-2 Phylogenies than de novo and Maximum-Likelihood Implementations.Syst Biol. 2023 Nov 1;72(5):1039-1051. doi: 10.1093/sysbio/syad031. Syst Biol. 2023. PMID: 37232476 Free PMC article.
-
A systematic review comparing at-home diagnostic tests for SARS-CoV-2: Key points for pharmacy practice, including regulatory information.J Am Pharm Assoc (2003). 2021 Nov-Dec;61(6):666-677.e2. doi: 10.1016/j.japh.2021.06.012. Epub 2021 Jun 12. J Am Pharm Assoc (2003). 2021. PMID: 34274214 Free PMC article.
-
Replacement of SARS-CoV-2 strains with variants carrying N501Y and L452R mutations in Japan: an epidemiological surveillance assessment.Western Pac Surveill Response J. 2022 Sep 16;13(3):1-10. doi: 10.5365/wpsar.2022.13.3.943. eCollection 2022 Jul-Sep. Western Pac Surveill Response J. 2022. PMID: 36688179 Free PMC article.
-
SARS-CoV-2 variant exposures elicit antibody responses with differential cross-neutralization of established and emerging strains including Delta and Omicron.medRxiv [Preprint]. 2021 Dec 22:2021.09.08.21263095. doi: 10.1101/2021.09.08.21263095. medRxiv. 2021. Update in: J Infect Dis. 2022 Jun 1;225(11):1909-1914. doi: 10.1093/infdis/jiab635. PMID: 34981075 Free PMC article. Updated. Preprint.
-
The biological and clinical significance of emerging SARS-CoV-2 variants.Nat Rev Genet. 2021 Dec;22(12):757-773. doi: 10.1038/s41576-021-00408-x. Epub 2021 Sep 17. Nat Rev Genet. 2021. PMID: 34535792 Free PMC article. Review.
References
-
- Voloch CM, Francisco R da S, Almeida LGP de, et al. . Genomic characterization of a novel SARS-CoV-2 lineage from Rio de Janeiro, Brazil. J Virol 2021; 95. Available at: https://jvi.asm.org/content/95/10/e00119-21. Accessed 17 May 2021. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
