

Normal and Premature Adrenarche
- PMID: 33788946
- PMCID: PMC8599200
- DOI: 10.1210/endrev/bnab009
Normal and Premature Adrenarche
Abstract
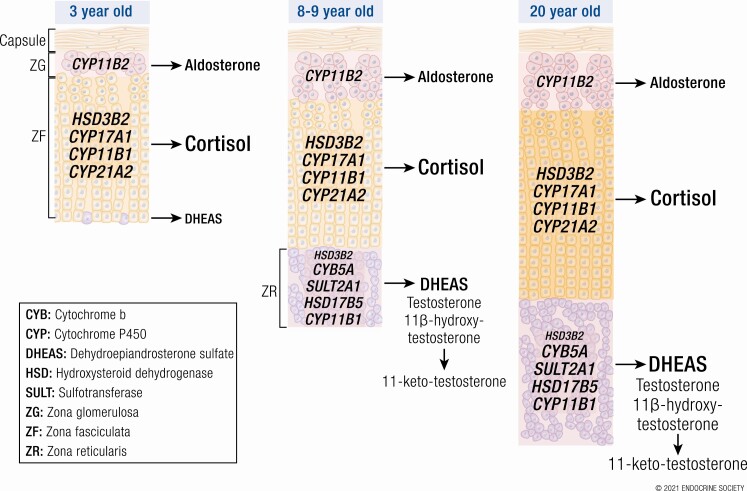
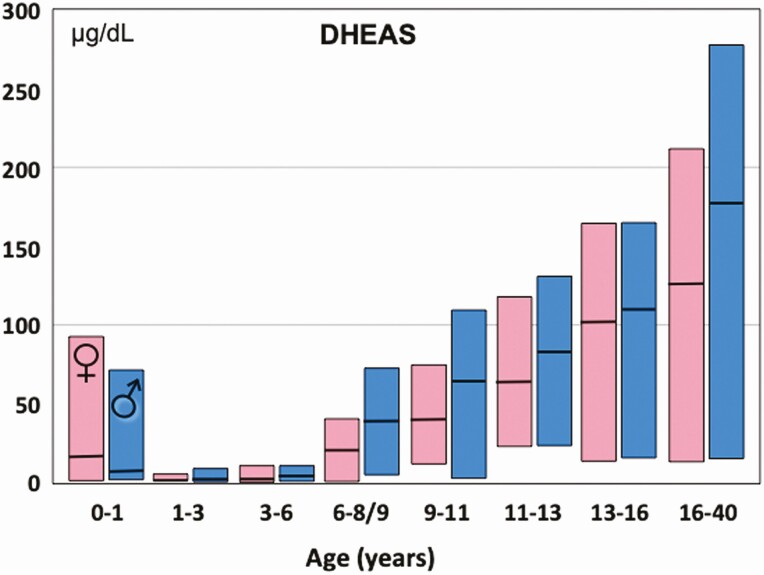
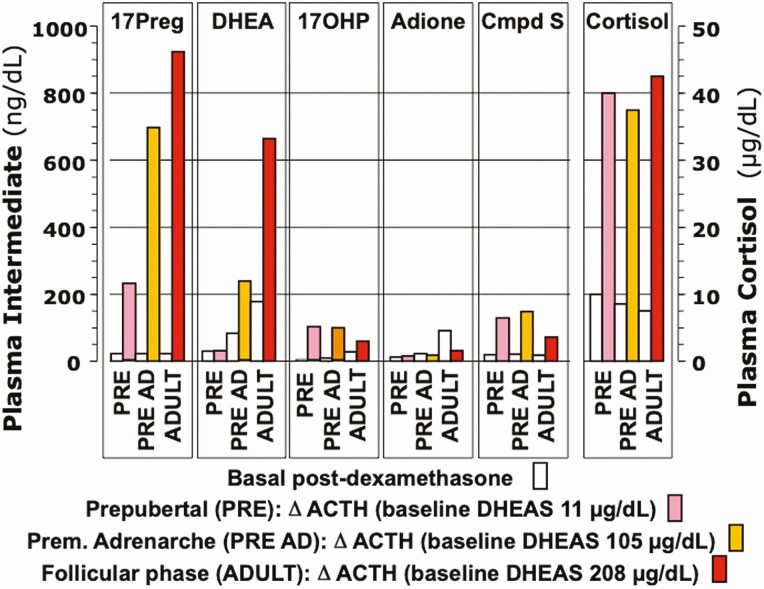
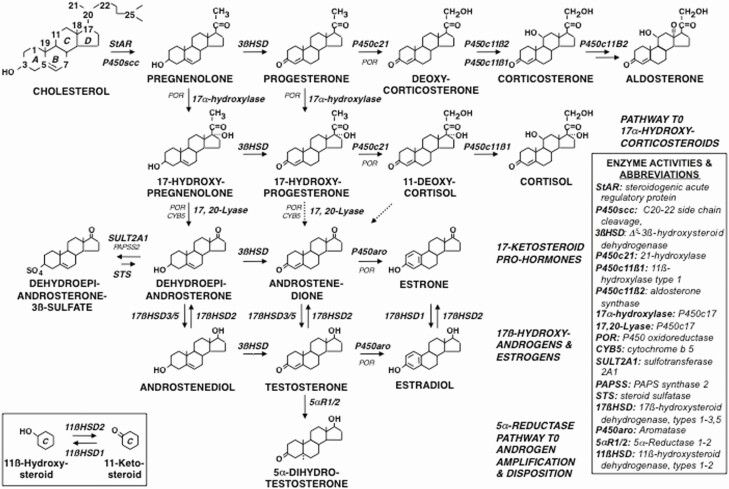
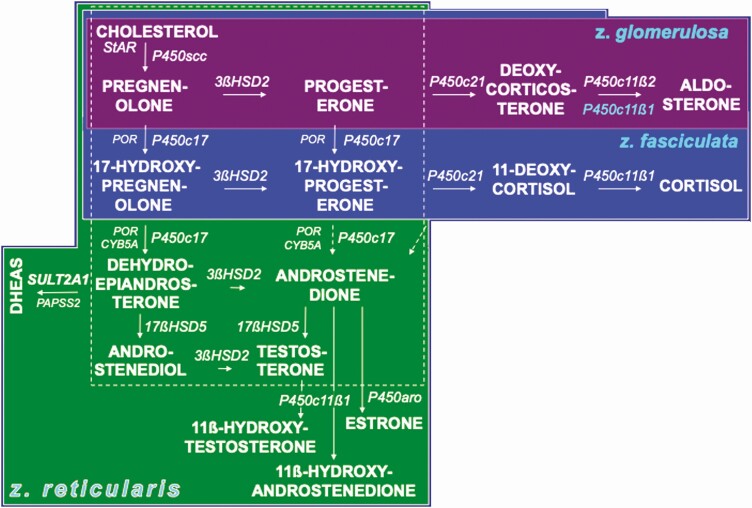
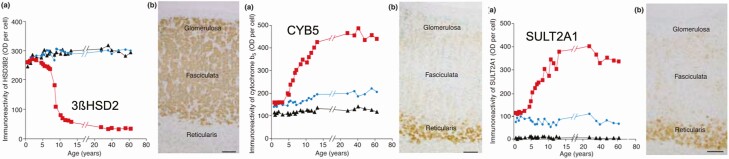
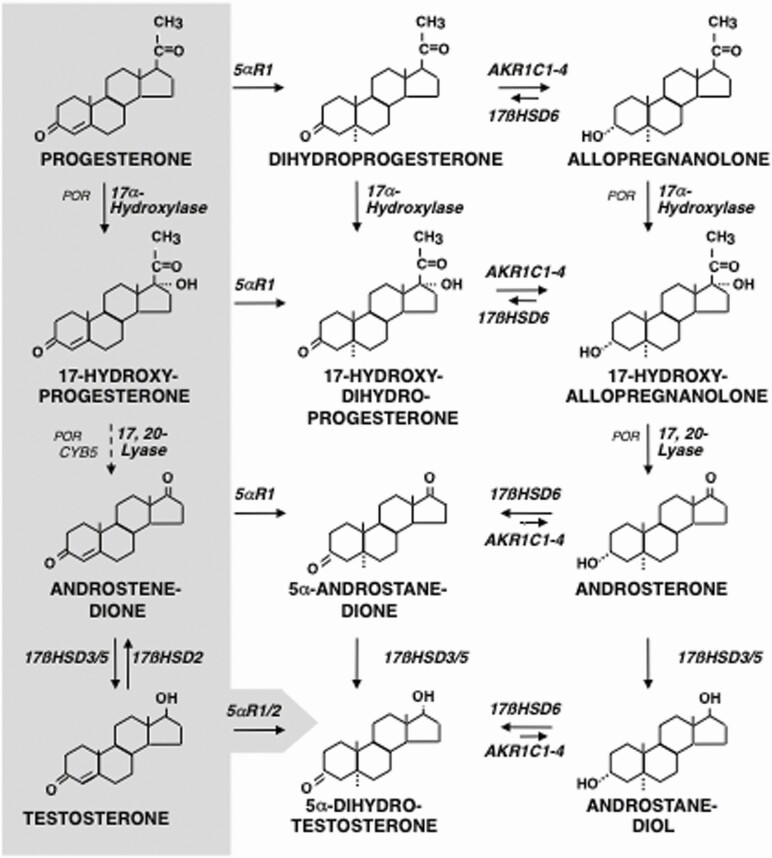
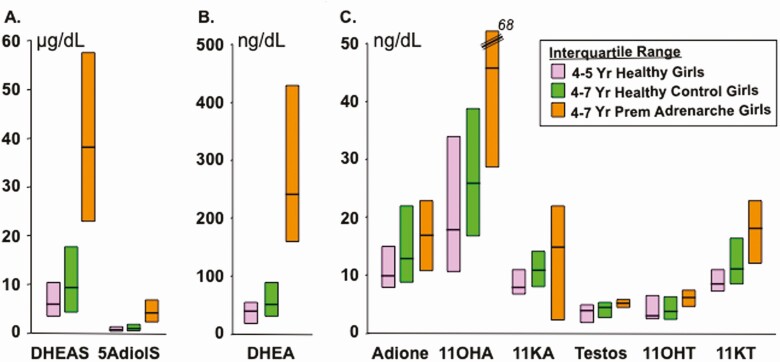
Adrenarche is the maturational increase in adrenal androgen production that normally begins in early childhood. It results from changes in the secretory response to adrenocorticotropin (ACTH) that are best indexed by dehydroepiandrosterone sulfate (DHEAS) rise. These changes are related to the development of the zona reticularis (ZR) and its unique gene/enzyme expression pattern of low 3ß-hydroxysteroid dehydrogenase type 2 with high cytochrome b5A, sulfotransferase 2A1, and 17ß-hydroxysteroid dehydrogenase type 5. Recently 11-ketotestosterone was identified as an important bioactive adrenarchal androgen. Birth weight, body growth, obesity, and prolactin are related to ZR development. Adrenarchal androgens normally contribute to the onset of sexual pubic hair (pubarche) and sebaceous and apocrine gland development. Premature adrenarche causes ≥90% of premature pubarche (PP). Its cause is unknown. Affected children have a significantly increased growth rate with proportionate bone age advancement that typically does not compromise growth potential. Serum DHEAS and testosterone levels increase to levels normal for early female puberty. It is associated with mildly increased risks for obesity, insulin resistance, and possibly mood disorder and polycystic ovary syndrome. Between 5% and 10% of PP is due to virilizing disorders, which are usually characterized by more rapid advancement of pubarche and compromise of adult height potential than premature adrenarche. Most cases are due to nonclassic congenital adrenal hyperplasia. Algorithms are presented for the differential diagnosis of PP. This review highlights recent advances in molecular genetic and developmental biologic understanding of ZR development and insights into adrenarche emanating from mass spectrometric steroid assays.
Keywords: adrenal androgens; adrenarche; polycystic ovary syndrome; pubarche; steroidogenic enzyme expression; zona reticularis.
© The Author(s) 2021. Published by Oxford University Press on behalf of the Endocrine Society. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
Figures
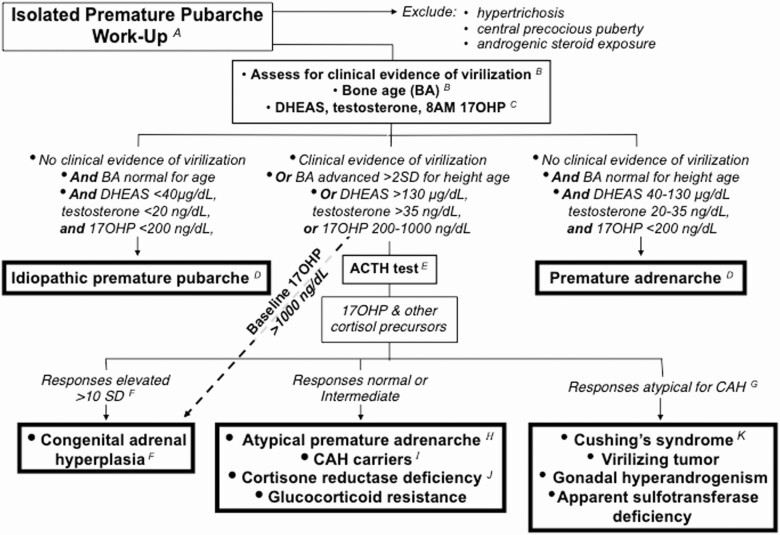
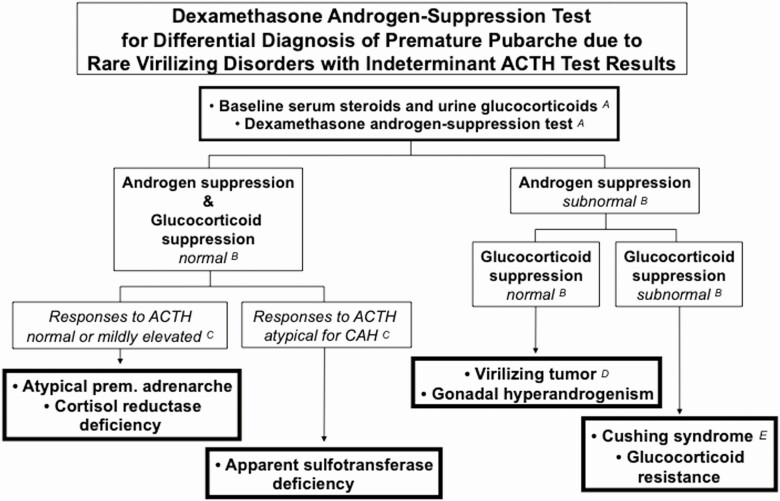
References
-
- Ibáñez L, Dimartino-Nardi J, Potau N, Saenger P. Premature adrenarche: normal variant or forerunner of adult disease? Endocr Rev. 2000;21(6):671-696. - PubMed
-
- Kulle AE, Reinehr T, Simic-Schleicher G, Hornig NC, Holterhus PM. Determination of 17OHPreg and DHEAS by LC-MS/MS: impact of age, sex, pubertal stage, and BMI on the Δ5 steroid pathway. J Clin Endocrinol Metab. 2017;102(1):232-241. - PubMed
-
- Rich BH, Rosenfield RL, Lucky AW, Helke JC, Otto P. Adrenarche: changing adrenal response to adrenocorticotropin. J Clin Endocrinol Metab. 1981;52(6):1129-1134. - PubMed
-
- Dhom G. The prepuberal and puberal growth of the adrenal (adrenarche). Beitr Pathol. 1973;150(4):357-377. - PubMed
-
- Suzuki T, Sasano H, Takeyama J, et al. Developmental changes in steroidogenic enzymes in human postnatal adrenal cortex: immunohistochemical studies. Clin Endocrinol (Oxf). 2000;53(6):739-747. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
