

Expectations and blind spots for structural variation detection from long-read assemblies and short-read genome sequencing technologies
- PMID: 33789087
- PMCID: PMC8206509
- DOI: 10.1016/j.ajhg.2021.03.014
Expectations and blind spots for structural variation detection from long-read assemblies and short-read genome sequencing technologies
Abstract
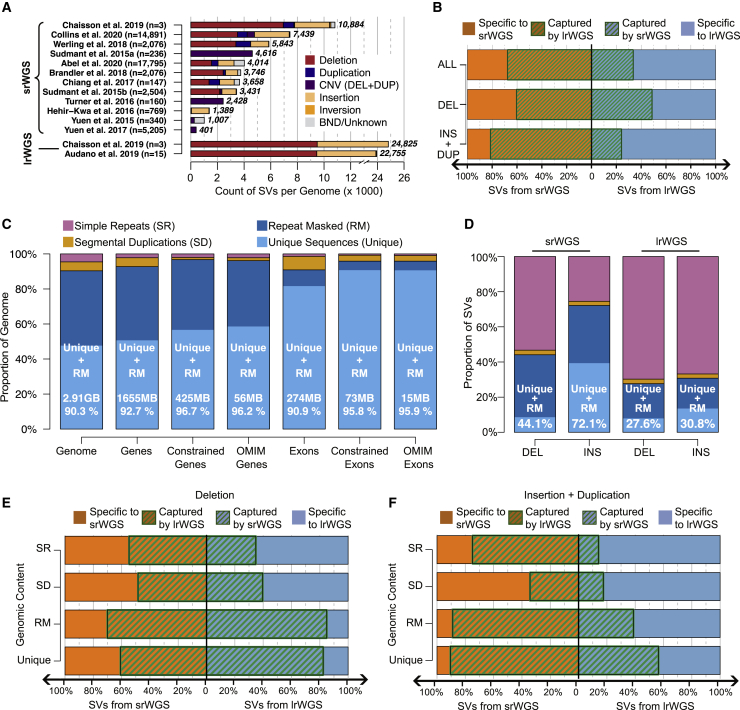
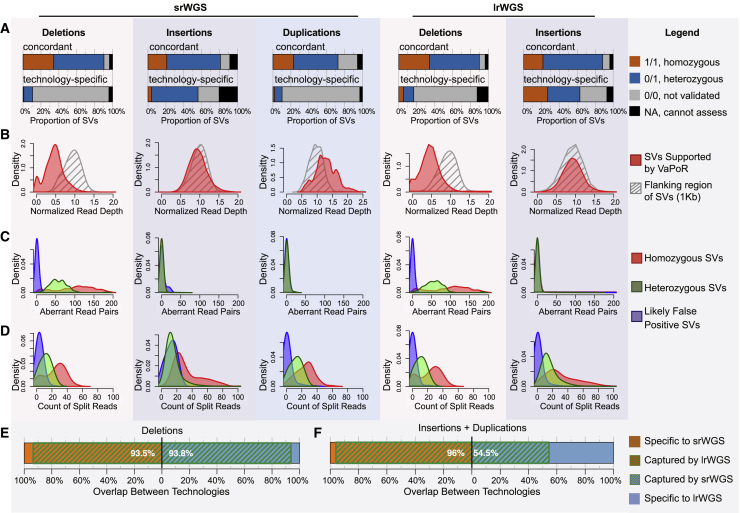
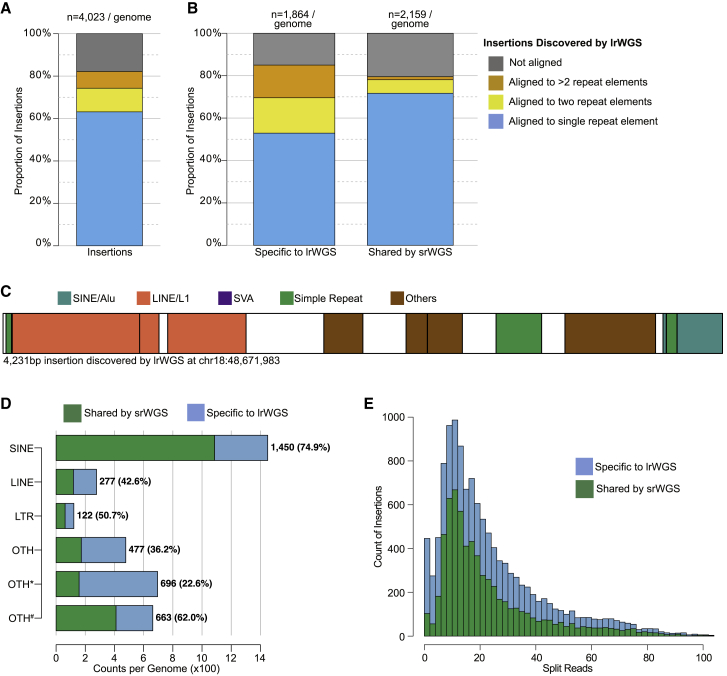
Virtually all genome sequencing efforts in national biobanks, complex and Mendelian disease programs, and medical genetic initiatives are reliant upon short-read whole-genome sequencing (srWGS), which presents challenges for the detection of structural variants (SVs) relative to emerging long-read WGS (lrWGS) technologies. Given this ubiquity of srWGS in large-scale genomics initiatives, we sought to establish expectations for routine SV detection from this data type by comparison with lrWGS assembly, as well as to quantify the genomic properties and added value of SVs uniquely accessible to each technology. Analyses from the Human Genome Structural Variation Consortium (HGSVC) of three families captured ~11,000 SVs per genome from srWGS and ~25,000 SVs per genome from lrWGS assembly. Detection power and precision for SV discovery varied dramatically by genomic context and variant class: 9.7% of the current GRCh38 reference is defined by segmental duplication (SD) and simple repeat (SR), yet 91.4% of deletions that were specifically discovered by lrWGS localized to these regions. Across the remaining 90.3% of reference sequence, we observed extremely high (93.8%) concordance between technologies for deletions in these datasets. In contrast, lrWGS was superior for detection of insertions across all genomic contexts. Given that non-SD/SR sequences encompass 95.9% of currently annotated disease-associated exons, improved sensitivity from lrWGS to discover novel pathogenic deletions in these currently interpretable genomic regions is likely to be incremental. However, these analyses highlight the considerable added value of assembly-based lrWGS to create new catalogs of insertions and transposable elements, as well as disease-associated repeat expansions in genomic sequences that were previously recalcitrant to routine assessment.
Keywords: copy number variation; genome assembly; long-read technology; segmental duplication; simple repeats; structural variation; whole-genome sequencing.
Copyright © 2021. Published by Elsevier Inc.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Posey J.E., O’Donnell-Luria A.H., Chong J.X., Harel T., Jhangiani S.N., Coban Akdemir Z.H., Buyske S., Pehlivan D., Carvalho C.M.B., Baxter S., Centers for Mendelian Genomics Insights into genetics, human biology and disease gleaned from family based genomic studies. Genet. Med. 2019;21:798–812. - PMC - PubMed
-
- Wright C.F., Fitzgerald T.W., Jones W.D., Clayton S., McRae J.F., van Kogelenberg M., King D.A., Ambridge K., Barrett D.M., Bayzetinova T., DDD study Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet. 2015;385:1305–1314. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
- R01 HG002898/HG/NHGRI NIH HHS/United States
- R01 MH115957/MH/NIMH NIH HHS/United States
- R03 HD099547/HD/NICHD NIH HHS/United States
- R35 GM138212/GM/NIGMS NIH HHS/United States
- UM1 HG008895/HG/NHGRI NIH HHS/United States
- F31 HG010569/HG/NHGRI NIH HHS/United States
- T32 HG002295/HG/NHGRI NIH HHS/United States
- R01 HG010169/HG/NHGRI NIH HHS/United States
- R01 HD081256/HD/NICHD NIH HHS/United States
- U24 HG007497/HG/NHGRI NIH HHS/United States
- R01 HD096326/HD/NICHD NIH HHS/United States
- R00 DE026824/DE/NIDCR NIH HHS/United States
- P30 CA034196/CA/NCI NIH HHS/United States
- T32 HG000040/HG/NHGRI NIH HHS/United States
- R01 HD091797/HD/NICHD NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
