

Phosphodiesterase 1C integrates store-operated calcium entry and cAMP signaling in leading-edge protrusions of migrating human arterial myocytes
- PMID: 33789162
- PMCID: PMC8095186
- DOI: 10.1016/j.jbc.2021.100606
Phosphodiesterase 1C integrates store-operated calcium entry and cAMP signaling in leading-edge protrusions of migrating human arterial myocytes
Abstract
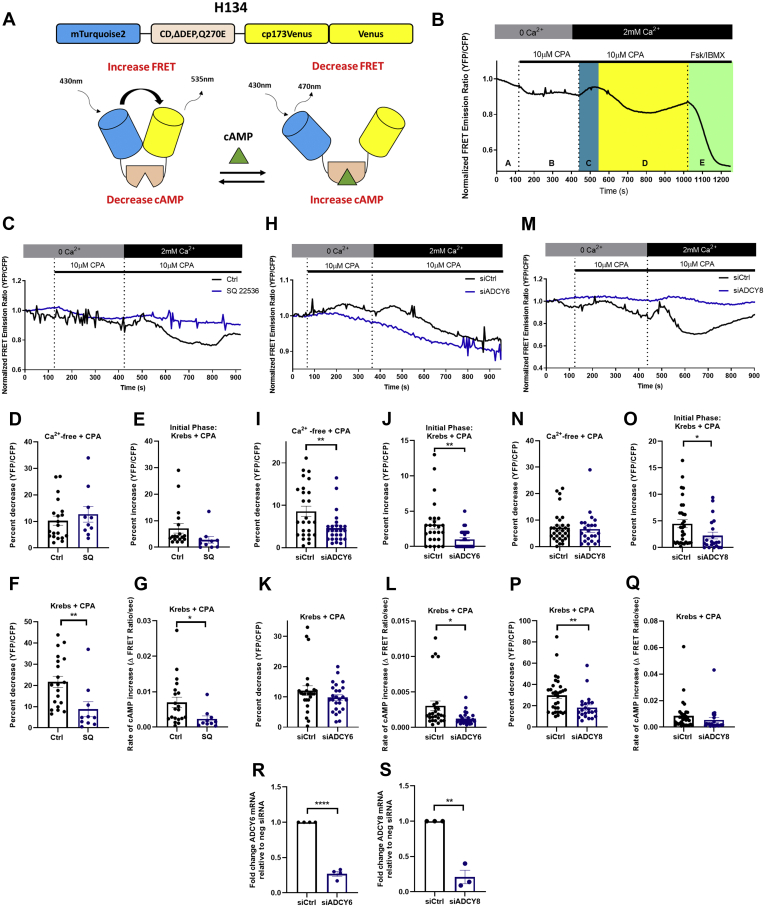
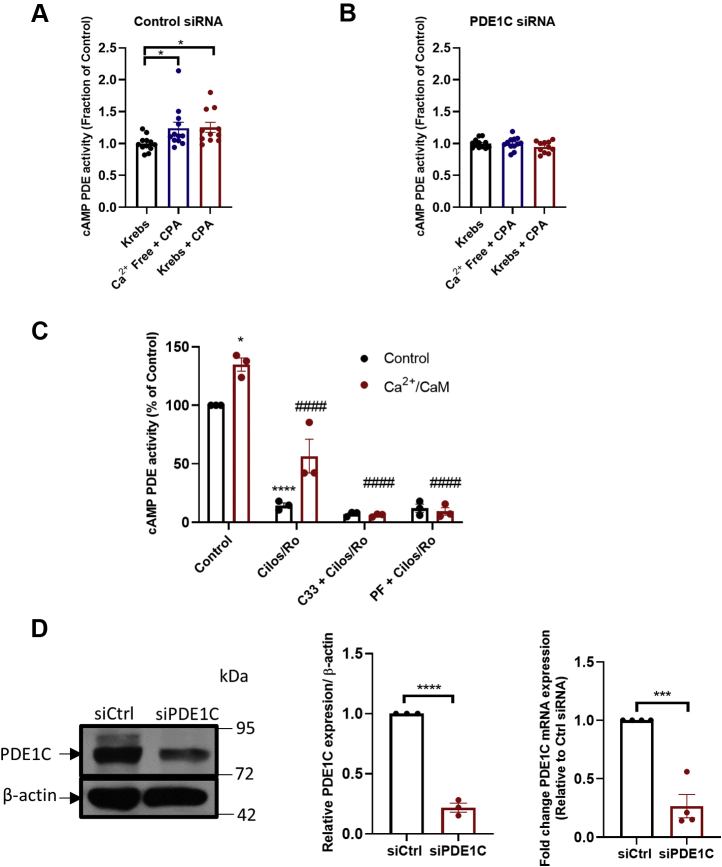
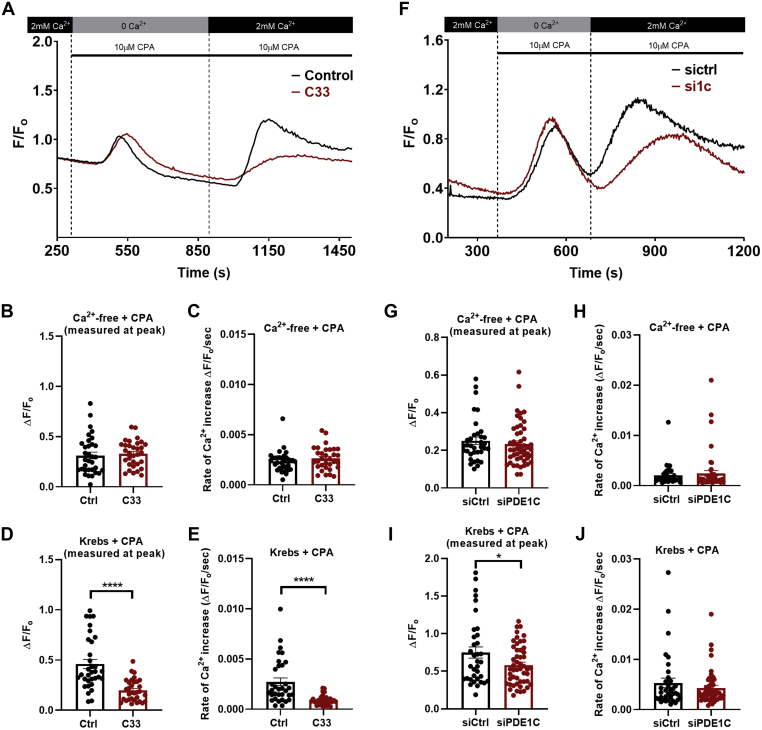
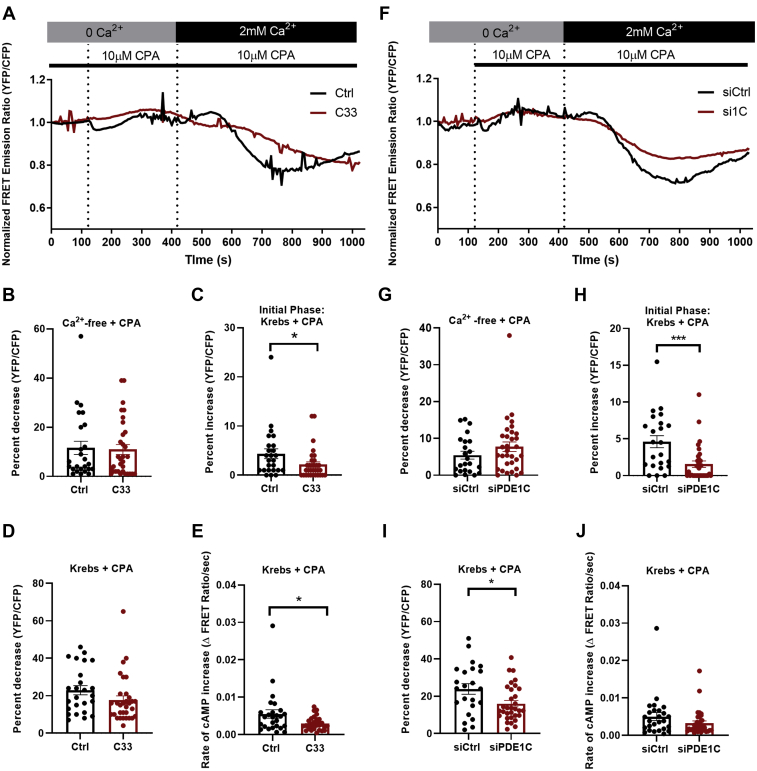
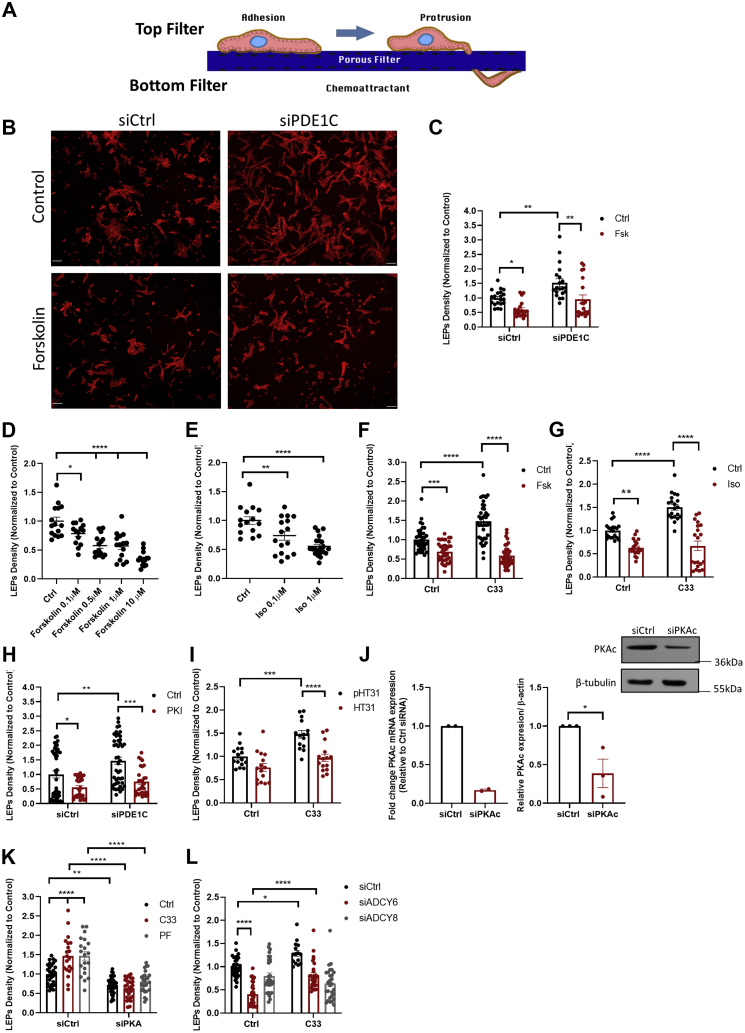
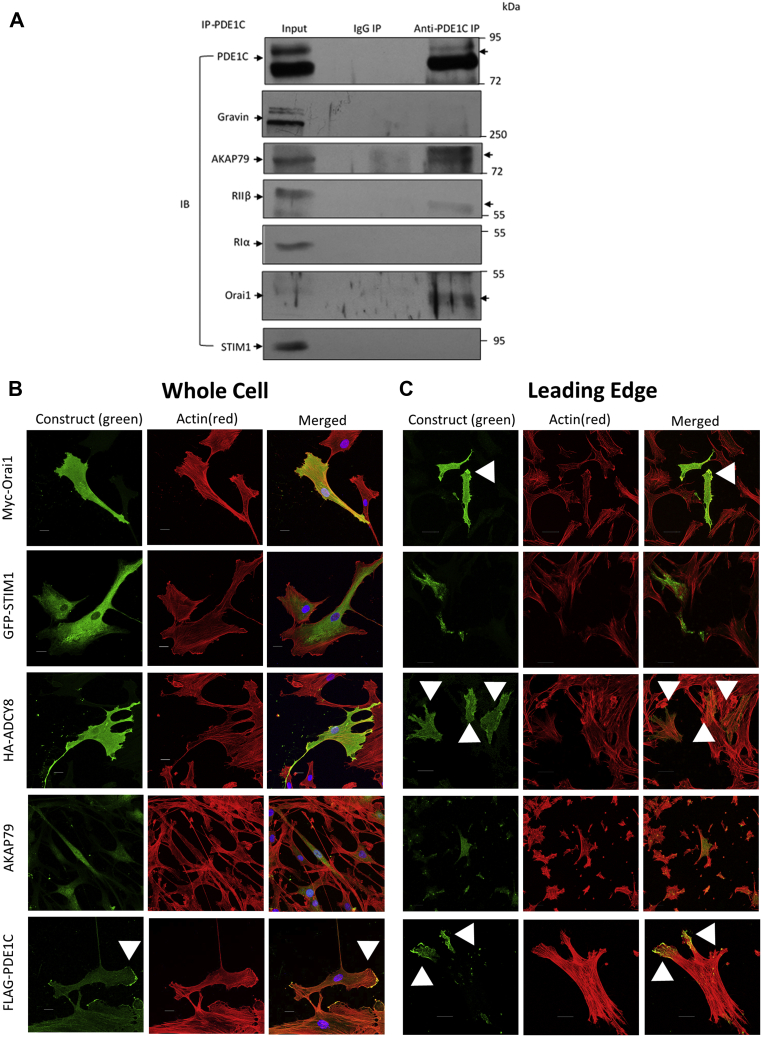
In addition to maintaining cellular ER Ca2+ stores, store-operated Ca2+ entry (SOCE) regulates several Ca2+-sensitive cellular enzymes, including certain adenylyl cyclases (ADCYs), enzymes that synthesize the secondary messenger cyclic AMP (cAMP). Ca2+, acting with calmodulin, can also increase the activity of PDE1-family phosphodiesterases (PDEs), which cleave the phosphodiester bond of cAMP. Surprisingly, SOCE-regulated cAMP signaling has not been studied in cells expressing both Ca2+-sensitive enzymes. Here, we report that depletion of ER Ca2+ activates PDE1C in human arterial smooth muscle cells (HASMCs). Inhibiting the activation of PDE1C reduced the magnitude of both SOCE and subsequent Ca2+/calmodulin-mediated activation of ADCY8 in these cells. Because inhibiting or silencing Ca2+-insensitive PDEs had no such effects, these data identify PDE1C-mediated hydrolysis of cAMP as a novel and important link between SOCE and its activation of ADCY8. Functionally, we showed that PDE1C regulated the formation of leading-edge protrusions in HASMCs, a critical early event in cell migration. Indeed, we found that PDE1C populated the tips of newly forming leading-edge protrusions in polarized HASMCs, and co-localized with ADCY8, the Ca2+ release activated Ca2+ channel subunit, Orai1, the cAMP-effector, protein kinase A, and an A-kinase anchoring protein, AKAP79. Because this polarization could allow PDE1C to control cAMP signaling in a hyper-localized manner, we suggest that PDE1C-selective therapeutic agents could offer increased spatial specificity in HASMCs over agents that regulate cAMP globally in cells. Similarly, such agents could also prove useful in regulating crosstalk between Ca2+/cAMP signaling in other cells in which dysregulated migration contributes to human pathology, including certain cancers.
Keywords: adenylate cyclase; calcium; cyclic AMP; human arterial smooth muscle cells; phosphodiesterase 1C; phosphodiesterases; store-operated calcium entry.
Copyright © 2021 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest None declared.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
