

The genetic architecture of human complex phenotypes is modulated by linkage disequilibrium and heterozygosity
- PMID: 33789345
- PMCID: PMC8045737
- DOI: 10.1093/genetics/iyaa046
The genetic architecture of human complex phenotypes is modulated by linkage disequilibrium and heterozygosity
Erratum in
-
Erratum to: The genetic architecture of human complex phenotypes is modulated by linkage disequilibrium and heterozygosity.Genetics. 2021 Jun 24;218(2):iyab064. doi: 10.1093/genetics/iyab064. Genetics. 2021. PMID: 34167151 Free PMC article. No abstract available.
Abstract
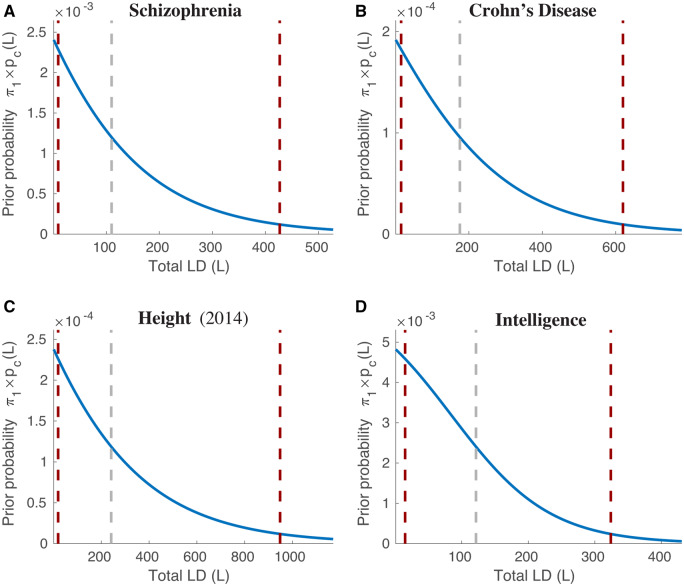
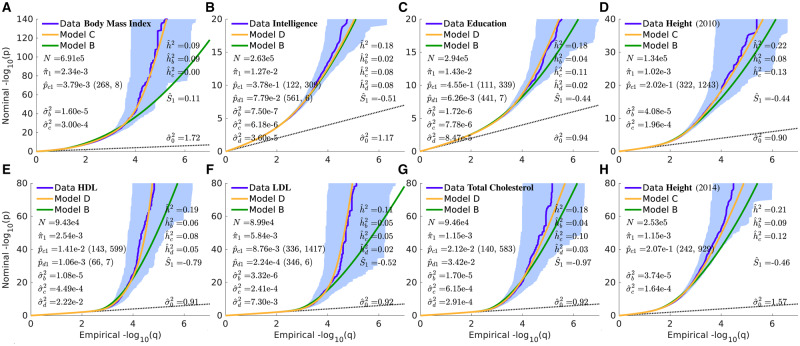
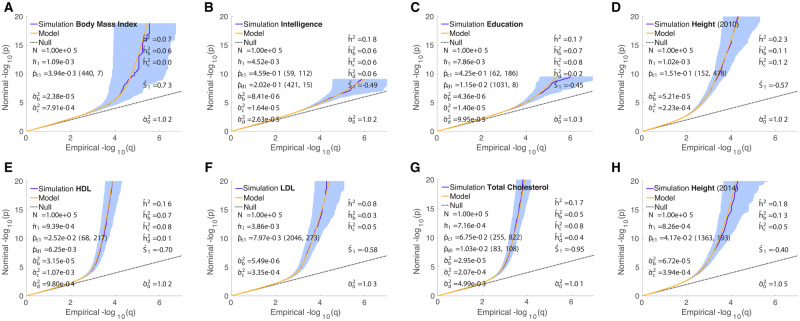
We propose an extended Gaussian mixture model for the distribution of causal effects of common single nucleotide polymorphisms (SNPs) for human complex phenotypes that depends on linkage disequilibrium (LD) and heterozygosity (H), while also allowing for independent components for small and large effects. Using a precise methodology showing how genome-wide association studies (GWASs) summary statistics (z-scores) arise through LD with underlying causal SNPs, we applied the model to GWAS of multiple human phenotypes. Our findings indicated that causal effects are distributed with dependence on total LD and H, whereby SNPs with lower total LD and H are more likely to be causal with larger effects; this dependence is consistent with models of the influence of negative pressure from natural selection. Compared with the basic Gaussian mixture model it is built on, the extended model-primarily through quantification of selection pressure-reproduces with greater accuracy the empirical distributions of z-scores, thus providing better estimates of genetic quantities, such as polygenicity and heritability, that arise from the distribution of causal effects.
Keywords: effect size; heritability; linkage disequilibrium; minor allele frequency; natural selection; polygenicity.
© The Author(s) 2021. Published by Oxford University Press on behalf of Genetics Society of America. All rights reserved. For permissions, please email: journals.permissions@oup.com.
Figures




References
-
- Alzheimer’s Association. 2018. 2018 Alzheimer’s disease facts and figures. Alzheimer’s Dementia, 14:367–429.
-
- Burisch J, Jess T, Martinato M, Lakatos PL, ECCO EpiCom.. 2013. The burden of inflammatory bowel disease in Europe. J Crohns Colitis. 7:322–337. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
