

Increasing brain palmitoylation rescues behavior and neuropathology in Huntington disease mice
- PMID: 33789888
- PMCID: PMC8011966
- DOI: 10.1126/sciadv.abb0799
Increasing brain palmitoylation rescues behavior and neuropathology in Huntington disease mice
Abstract
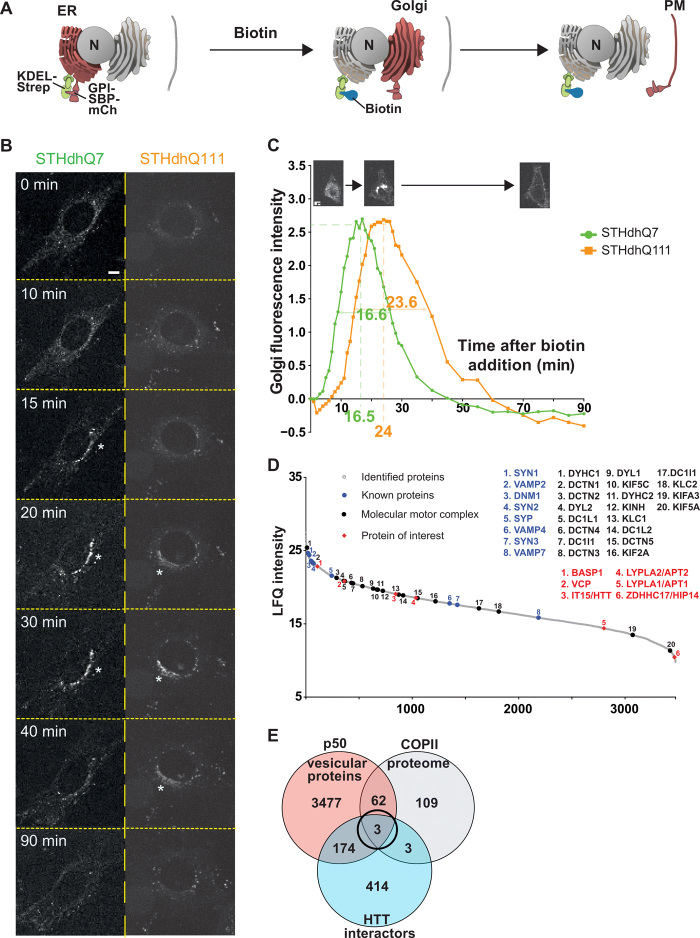
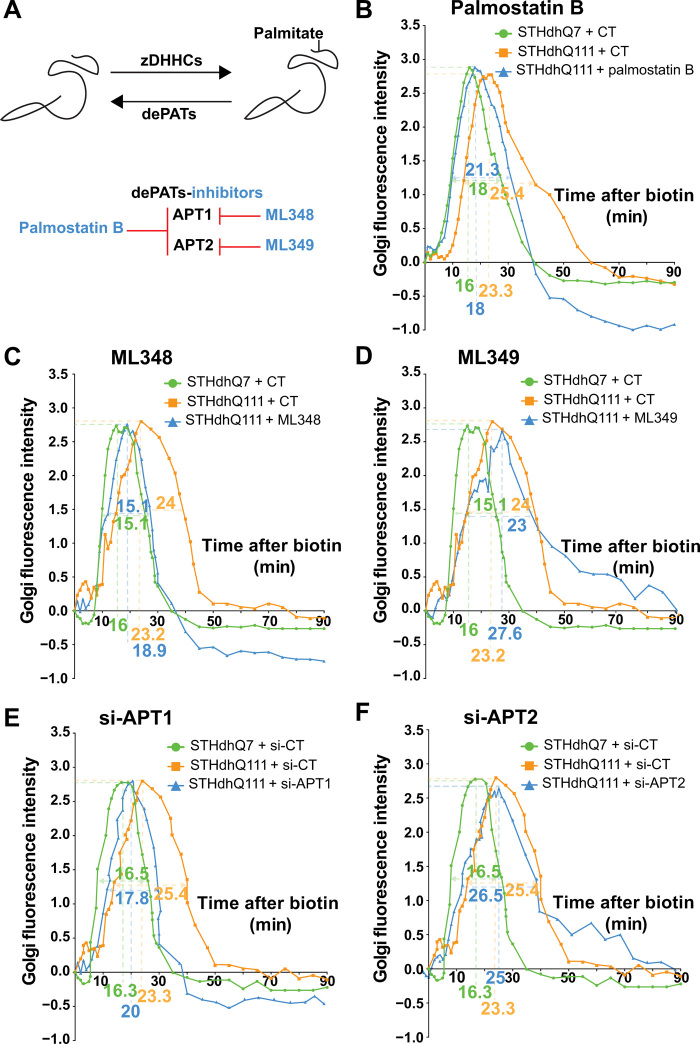
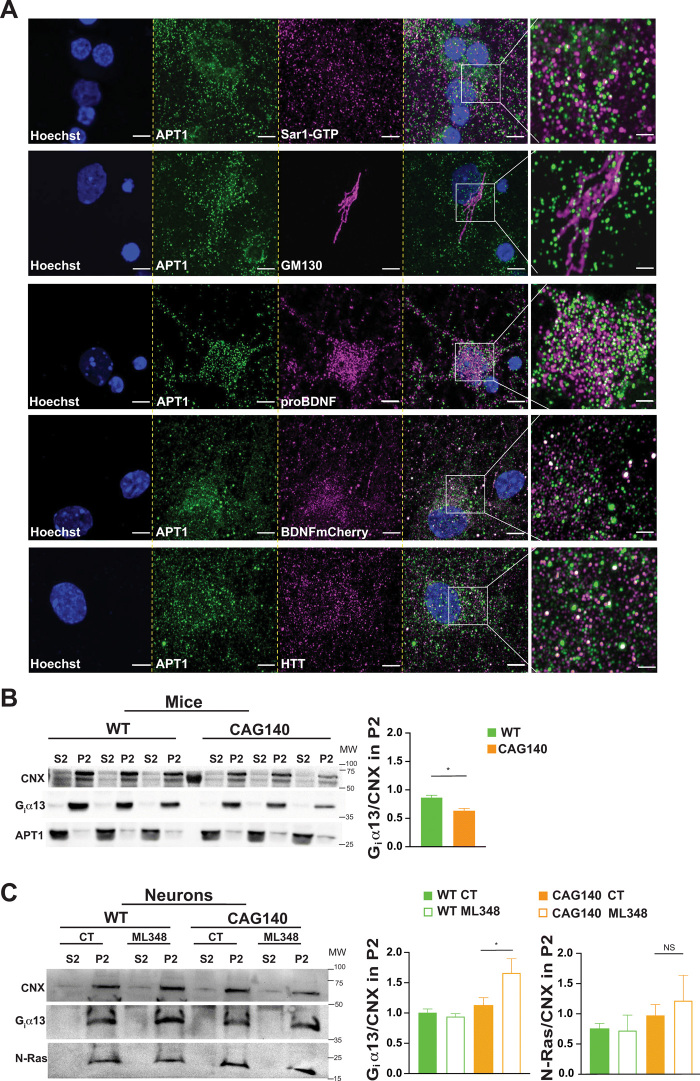
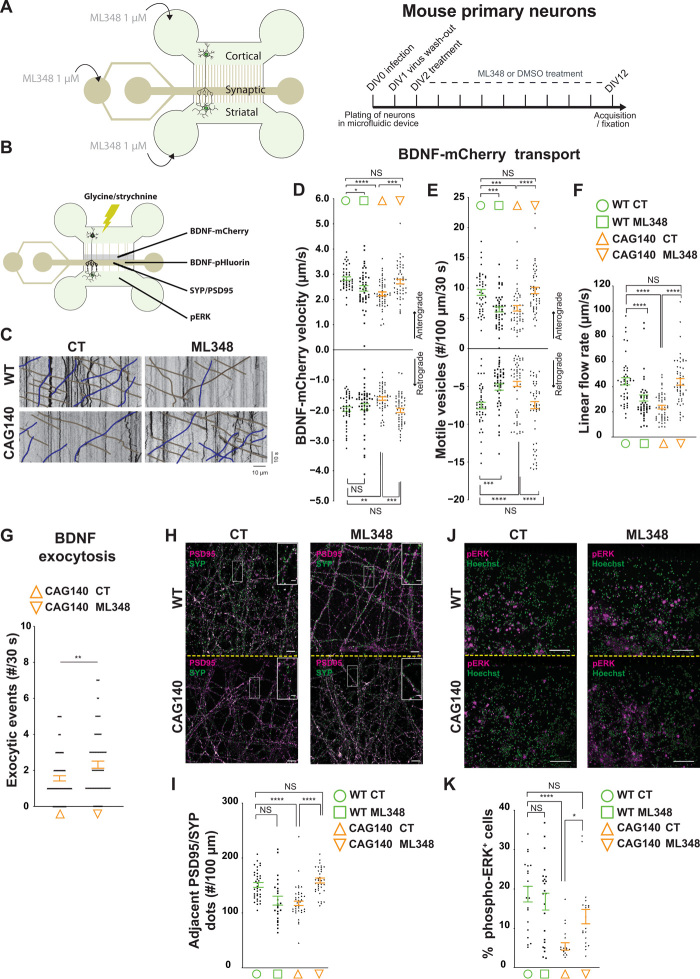
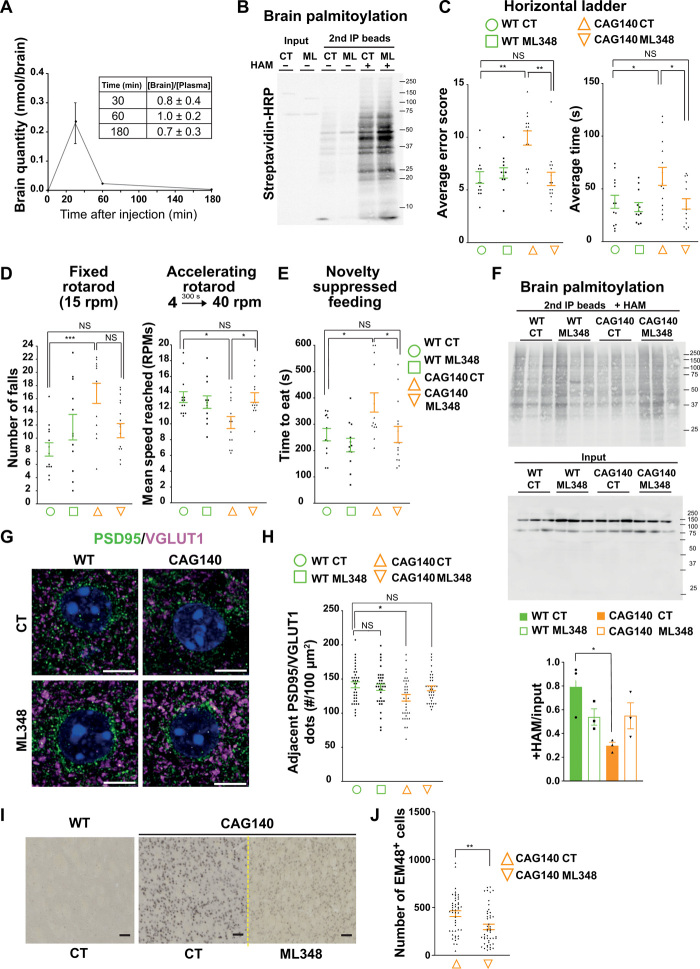
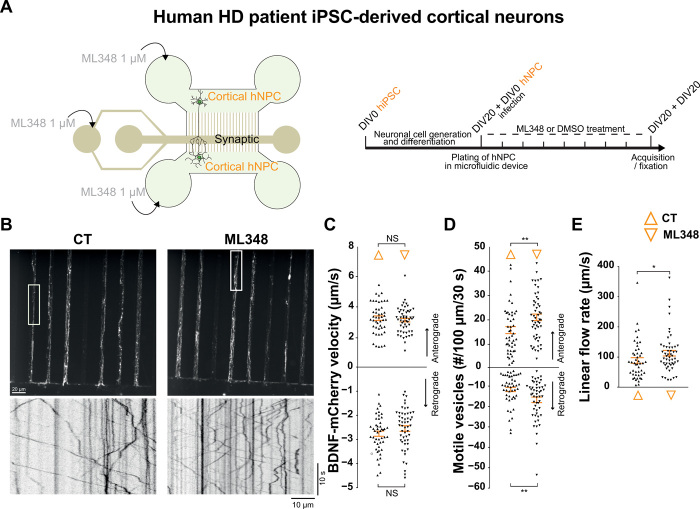
Huntington disease (HD) damages the corticostriatal circuitry in large part by impairing transport of brain-derived neurotrophic factor (BDNF). We hypothesized that improving vesicular transport of BDNF could slow or prevent disease progression. We therefore performed selective proteomic analysis of vesicles transported within corticostriatal projecting neurons followed by in silico screening and identified palmitoylation as a pathway that could restore defective huntingtin-dependent trafficking. Using a synchronized trafficking assay and an HD network-on-a-chip, we found that increasing brain palmitoylation via ML348, which inhibits the palmitate-removing enzyme acyl-protein thioesterase 1 (APT1), restores axonal transport, synapse homeostasis, and survival signaling to wild-type levels without toxicity. In human HD induced pluripotent stem cell-derived cortical neurons, ML348 increased BDNF trafficking. In HD knock-in mice, it efficiently crossed the blood-brain barrier to restore palmitoylation levels and reverse neuropathology, locomotor deficits, and anxio-depressive behaviors. APT1 and its inhibitor ML348 thus hold therapeutic interest for HD.
Copyright © 2021 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works. Distributed under a Creative Commons Attribution NonCommercial License 4.0 (CC BY-NC).
Figures
References
-
- Ross C. A., Tabrizi S. J., Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. 10, 83–98 (2011). - PubMed
-
- Saudou F., Humbert S., The biology of huntingtin. Neuron 89, 910–926 (2016). - PubMed
-
- Langfelder P., Cantle J. P., Chatzopoulou D., Wang N., Gao F., Al-Ramahi I., Lu X. H., Ramos E. M., El-Zein K., Zhao Y., Deverasetty S., Tebbe A., Schaab C., Lavery D. J., Howland D., Kwak S., Botas J., Aaronson J. S., Rosinski J., Coppola G., Horvath S., Yang X. W., Integrated genomics and proteomics define huntingtin CAG length-dependent networks in mice. Nat. Neurosci. 19, 623–633 (2016). - PMC - PubMed
-
- Al-Ramahi I., Lu B., Di Paola S., Pang K., de Haro M., Peluso I., Gallego-Flores T., Malik N. T., Erikson K., Bleiberg B. A., Avalos M., Fan G., Rivers L. E., Laitman A. M., Diaz-Garcia J. R., Hild M., Palacino J., Liu Z., Medina D. L., Botas J., High-throughput functional analysis distinguishes pathogenic, nonpathogenic, and compensatory transcriptional changes in neurodegeneration. Cell Syst. 7, 28–40.e4 (2018). - PMC - PubMed
-
- Becanovic K., Norremolle A., Neal S. J., Kay C., Collins J. A., Arenillas D., Lilja T., Gaudenzi G., Manoharan S., Doty C. N., Beck J., Lahiri N., Portales-Casamar E., Warby S. C., Connolly C., De Souza R. A.; REGISTRY Investigators of the European Huntington’s Disease Network, Tabrizi S. J., Hermanson O., Langbehn D. R., Hayden M. R., Wasserman W. W., Leavitt B. R., A SNP in the HTT promoter alters NF-κB binding and is a bidirectional genetic modifier of Huntington disease. Nat. Neurosci. 18, 807–816 (2015). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
