

Connecting structure to function with the recovery of over 1000 high-quality metagenome-assembled genomes from activated sludge using long-read sequencing
- PMID: 33790294
- PMCID: PMC8012365
- DOI: 10.1038/s41467-021-22203-2
Connecting structure to function with the recovery of over 1000 high-quality metagenome-assembled genomes from activated sludge using long-read sequencing
Abstract
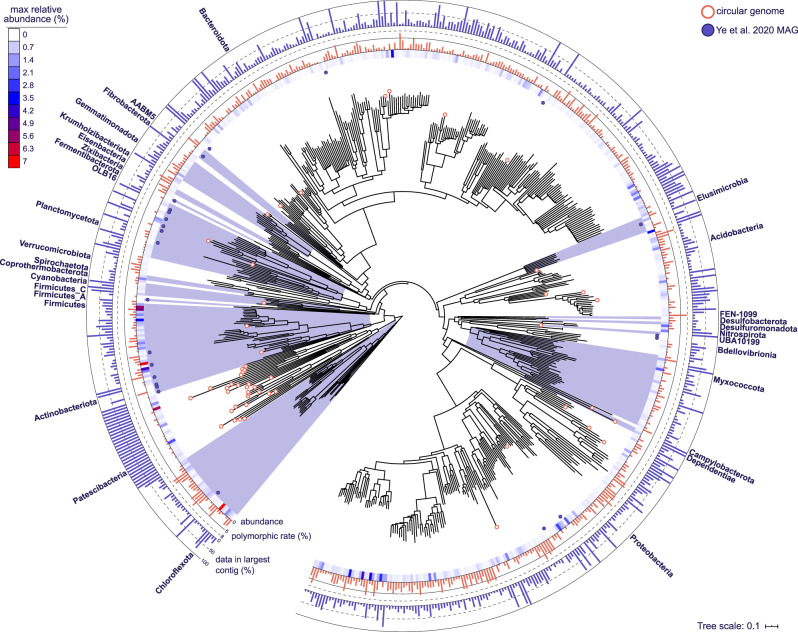
Microorganisms play crucial roles in water recycling, pollution removal and resource recovery in the wastewater industry. The structure of these microbial communities is increasingly understood based on 16S rRNA amplicon sequencing data. However, such data cannot be linked to functional potential in the absence of high-quality metagenome-assembled genomes (MAGs) for nearly all species. Here, we use long-read and short-read sequencing to recover 1083 high-quality MAGs, including 57 closed circular genomes, from 23 Danish full-scale wastewater treatment plants. The MAGs account for ~30% of the community based on relative abundance, and meet the stringent MIMAG high-quality draft requirements including full-length rRNA genes. We use the information provided by these MAGs in combination with >13 years of 16S rRNA amplicon sequencing data, as well as Raman microspectroscopy and fluorescence in situ hybridisation, to uncover abundant undescribed lineages belonging to important functional groups.
Conflict of interest statement
R.H.K., M.A., S.M.K., and P.H.N. own DNASense ApS. The remaining authors declare no competing interests.
Figures




References
Publication types
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
