

Age-dependent ataxia and neurodegeneration caused by an αII spectrin mutation with impaired regulation of its calpain sensitivity
- PMID: 33790315
- PMCID: PMC8012654
- DOI: 10.1038/s41598-021-86470-1
Age-dependent ataxia and neurodegeneration caused by an αII spectrin mutation with impaired regulation of its calpain sensitivity
Erratum in
-
Publisher Correction: Age-dependent ataxia and neurodegeneration caused by an αII spectrin mutation with impaired regulation of its calpain sensitivity.Sci Rep. 2021 Sep 8;11(1):18218. doi: 10.1038/s41598-021-97068-y. Sci Rep. 2021. PMID: 34497286 Free PMC article. No abstract available.
Abstract
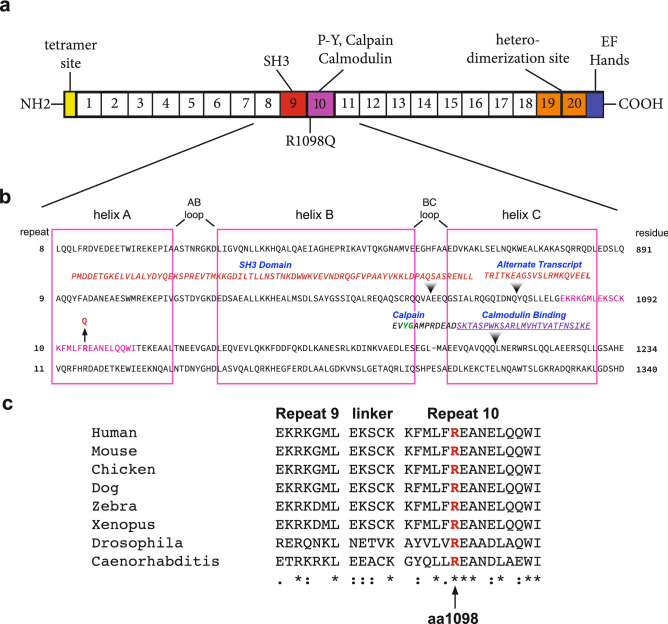
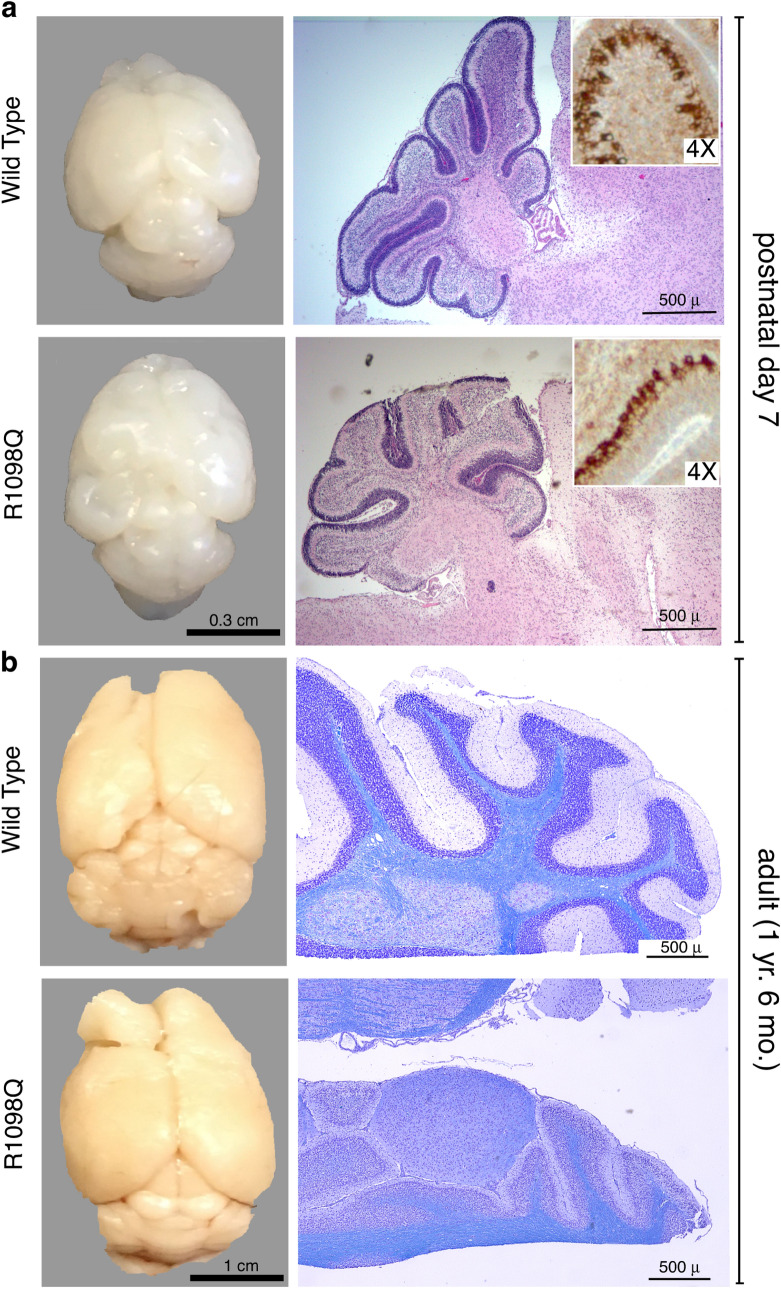
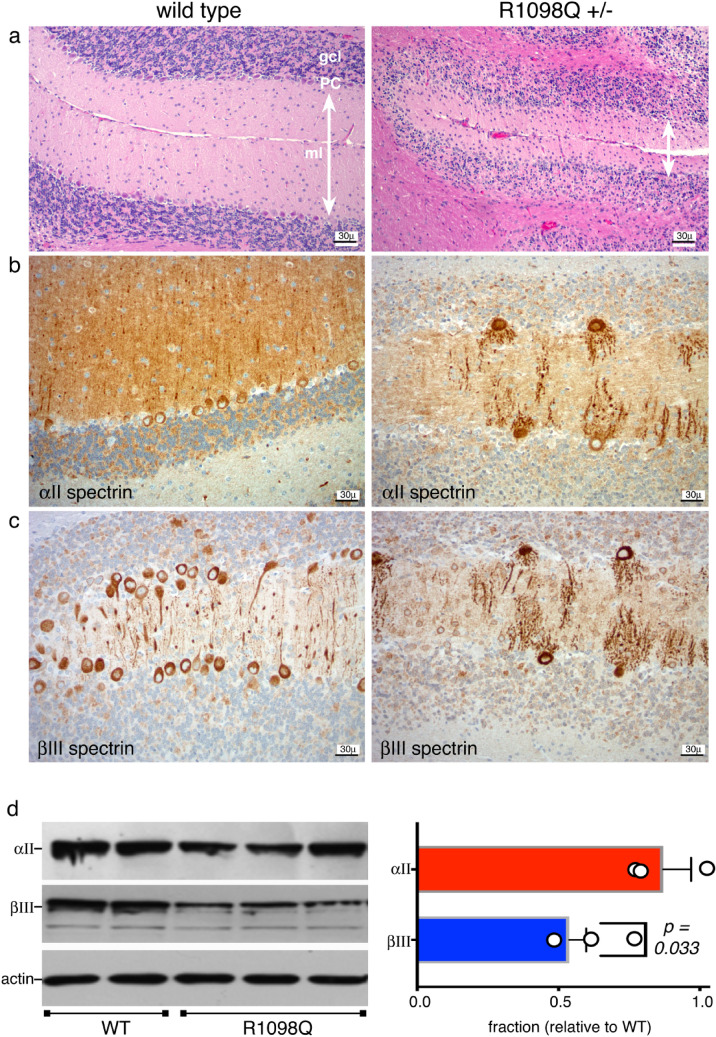
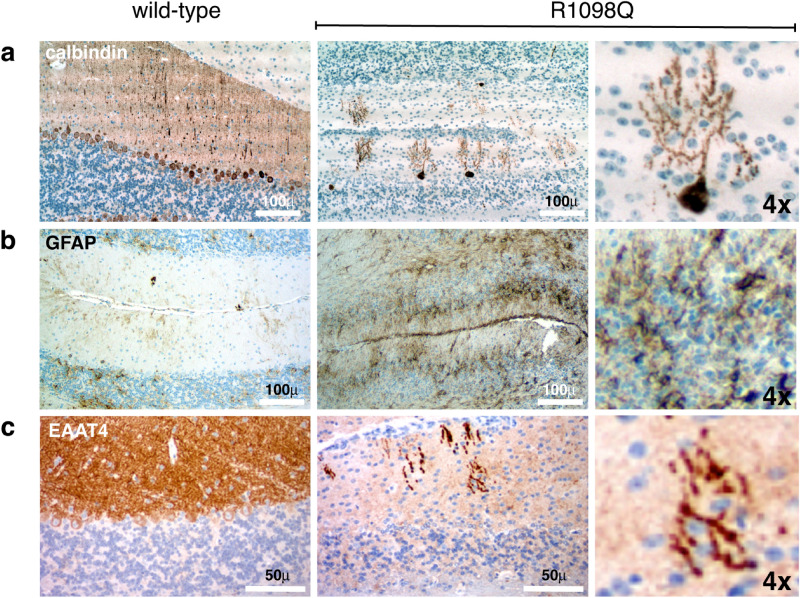
The neuronal membrane-associated periodic spectrin skeleton (MPS) contributes to neuronal development, remodeling, and organization. Post-translational modifications impinge on spectrin, the major component of the MPS, but their role remains poorly understood. One modification targeting spectrin is cleavage by calpains, a family of calcium-activated proteases. Spectrin cleavage is regulated by activated calpain, but also by the calcium-dependent binding of calmodulin (CaM) to spectrin. The physiologic significance of this balance between calpain activation and substrate-level regulation of spectrin cleavage is unknown. We report a strain of C57BL/6J mice harboring a single αII spectrin point mutation (Sptan1 c.3293G > A:p.R1098Q) with reduced CaM affinity and intrinsically enhanced sensitivity to calpain proteolysis. Homozygotes are embryonic lethal. Newborn heterozygotes of either gender appear normal, but soon develop a progressive ataxia characterized biochemically by accelerated calpain-mediated spectrin cleavage and morphologically by disruption of axonal and dendritic integrity and global neurodegeneration. Molecular modeling predicts unconstrained exposure of the mutant spectrin's calpain-cleavage site. These results reveal the critical importance of substrate-level regulation of spectrin cleavage for the maintenance of neuronal integrity. Given that excessive activation of calpain proteases is a common feature of neurodegenerative disease and traumatic encephalopathy, we propose that damage to the spectrin MPS may contribute to the neuropathology of many disorders.
Conflict of interest statement
The authors declare no competing interests.
Figures
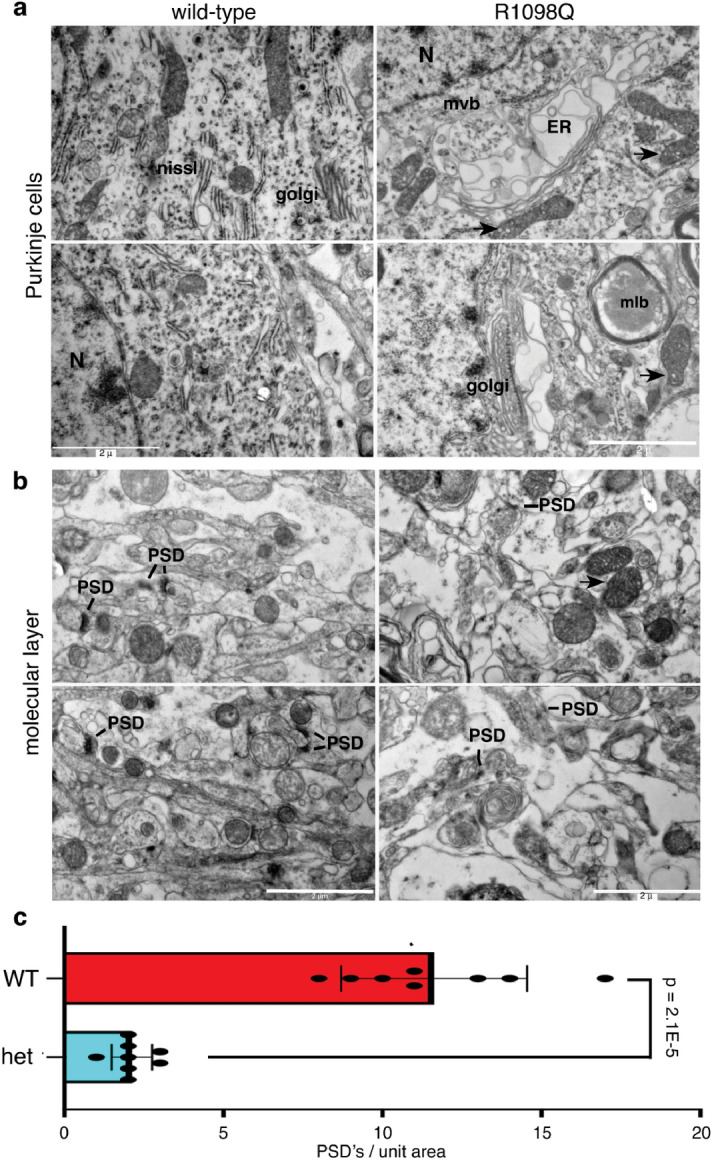
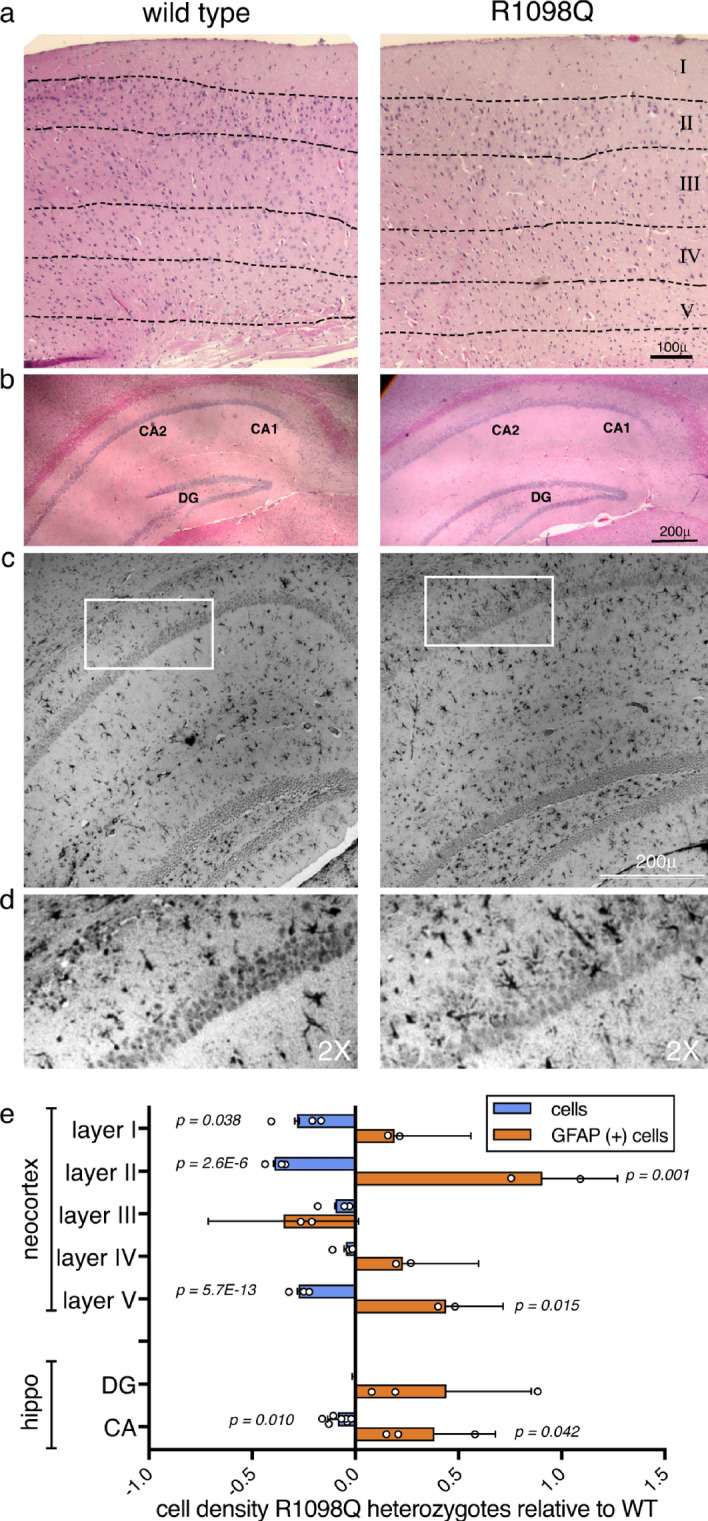
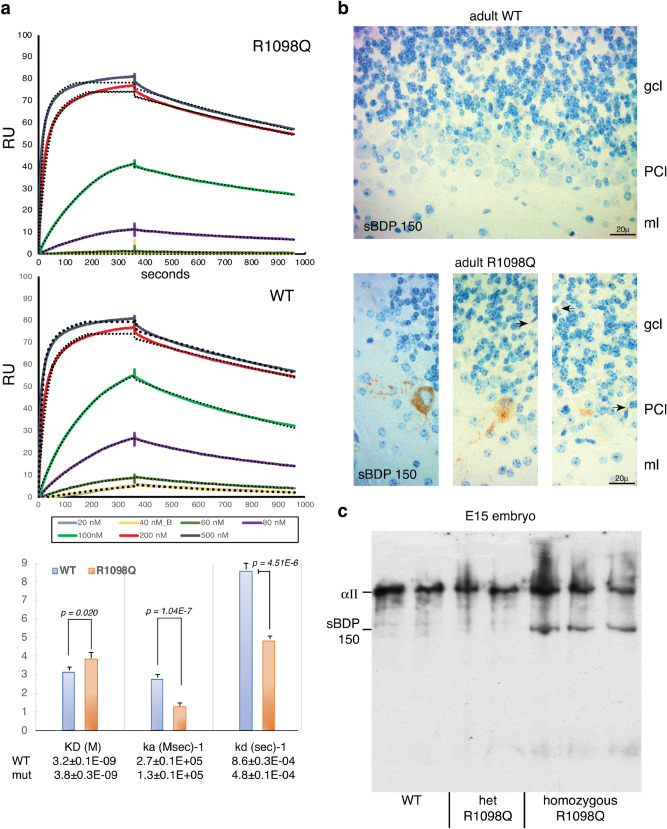
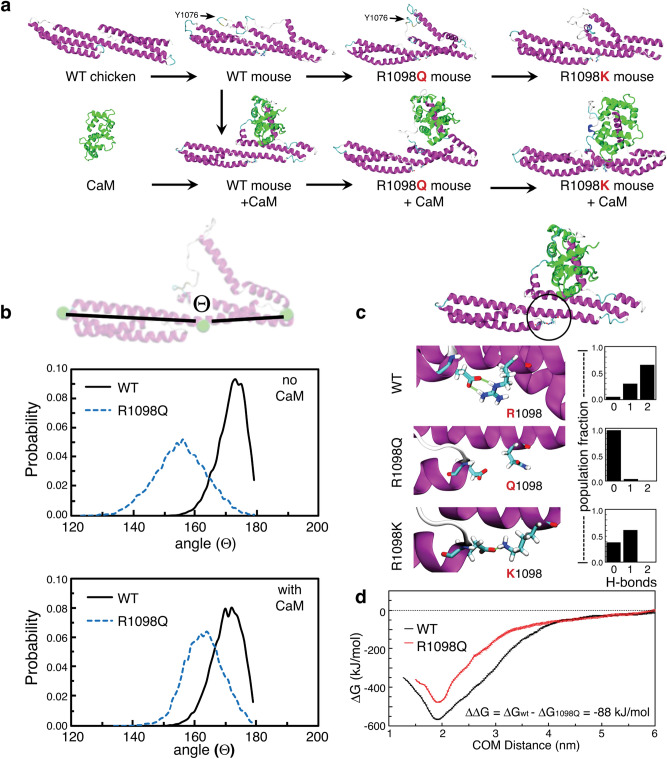
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
