

Emergence of Bat-Related Betacoronaviruses: Hazard and Risks
- PMID: 33790874
- PMCID: PMC8005542
- DOI: 10.3389/fmicb.2021.591535
Emergence of Bat-Related Betacoronaviruses: Hazard and Risks
Abstract
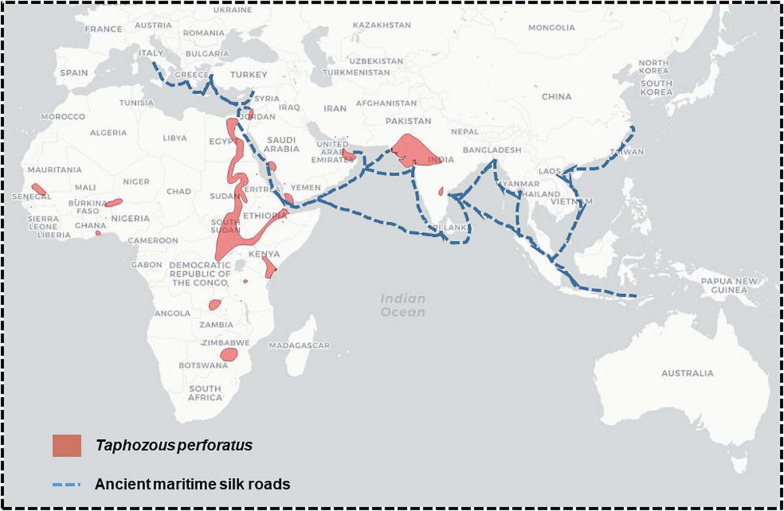
The current Coronavirus Disease 2019 (COVID-19) pandemic, with more than 111 million reported cases and 2,500,000 deaths worldwide (mortality rate currently estimated at 2.2%), is a stark reminder that coronaviruses (CoV)-induced diseases remain a major threat to humanity. COVID-19 is only the latest case of betacoronavirus (β-CoV) epidemics/pandemics. In the last 20 years, two deadly CoV epidemics, Severe Acute Respiratory Syndrome (SARS; fatality rate 9.6%) and Middle East Respiratory Syndrome (MERS; fatality rate 34.7%), plus the emergence of HCoV-HKU1 which causes the winter common cold (fatality rate 0.5%), were already a source of public health concern. Betacoronaviruses can also be a threat for livestock, as evidenced by the Swine Acute Diarrhea Syndrome (SADS) epizootic in pigs. These repeated outbreaks of β-CoV-induced diseases raise the question of the dynamic of propagation of this group of viruses in wildlife and human ecosystems. SARS-CoV, SARS-CoV-2, and HCoV-HKU1 emerged in Asia, strongly suggesting the existence of a regional hot spot for emergence. However, there might be other regional hot spots, as seen with MERS-CoV, which emerged in the Arabian Peninsula. β-CoVs responsible for human respiratory infections are closely related to bat-borne viruses. Bats are present worldwide and their level of infection with CoVs is very high on all continents. However, there is as yet no evidence of direct bat-to-human coronavirus infection. Transmission of β-CoV to humans is considered to occur accidentally through contact with susceptible intermediate animal species. This zoonotic emergence is a complex process involving not only bats, wildlife and natural ecosystems, but also many anthropogenic and societal aspects. Here, we try to understand why only few hot spots of β-CoV emergence have been identified despite worldwide bats and bat-borne β-CoV distribution. In this work, we analyze and compare the natural and anthropogenic environments associated with the emergence of β-CoV and outline conserved features likely to create favorable conditions for a new epidemic. We suggest monitoring South and East Africa as well as South America as these regions bring together many of the conditions that could make them future hot spots.
Keywords: COVID-19; MERS; SARS; coronavirus; hazard and risks assessment.
Copyright © 2021 Frutos, Serra-Cobo, Pinault, Lopez Roig and Devaux.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
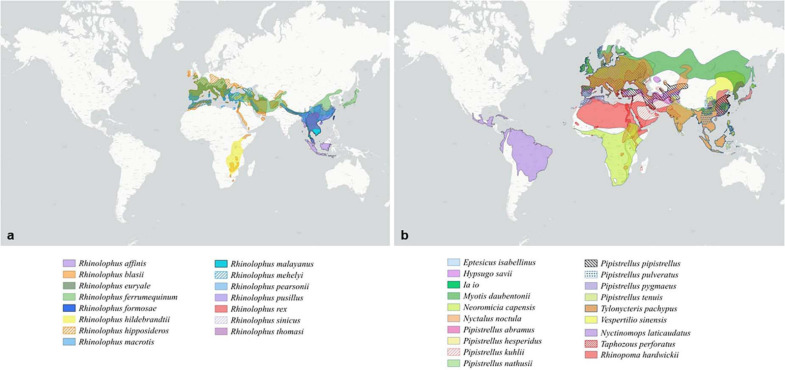
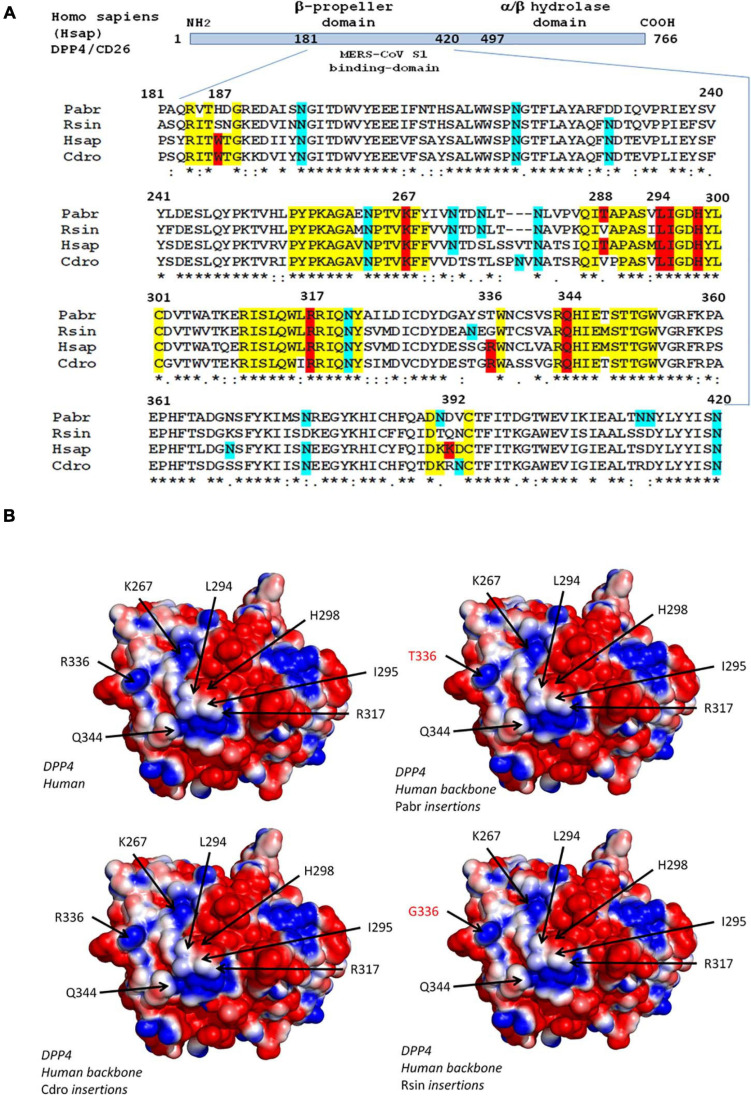
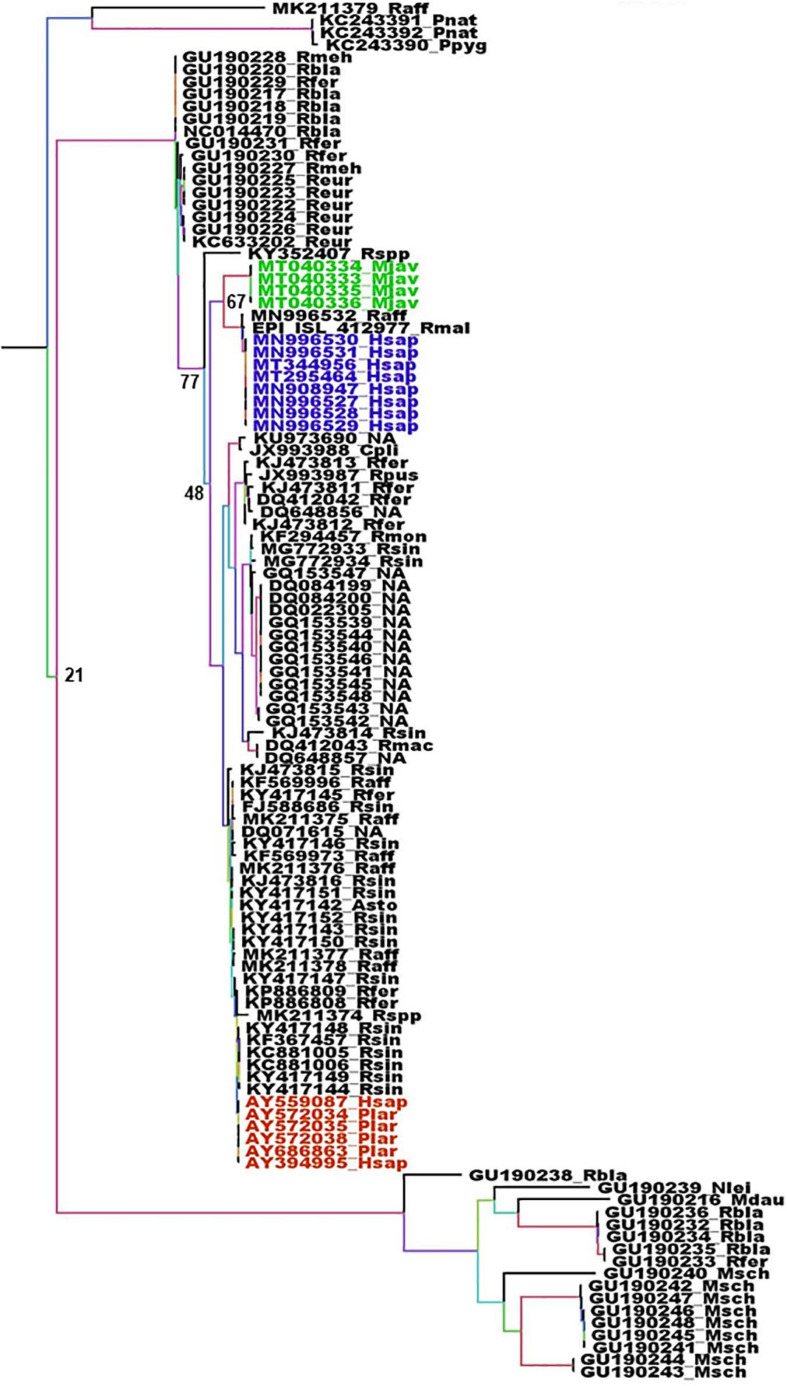
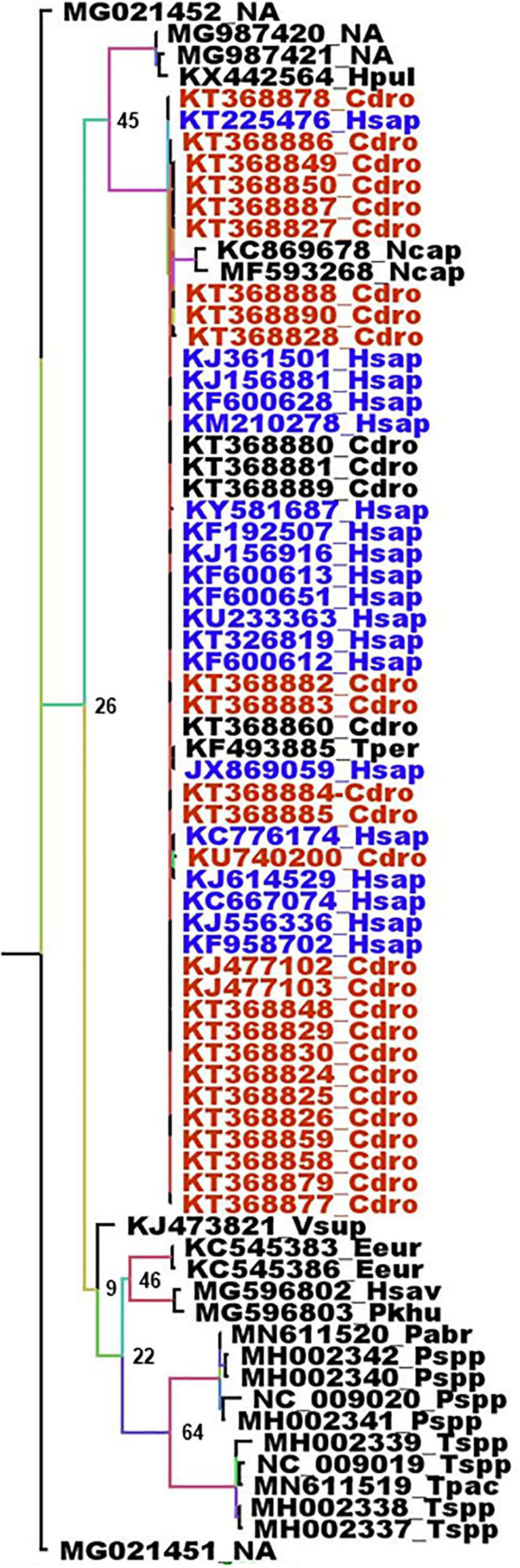
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous