

TMEM218 dysfunction causes ciliopathies, including Joubert and Meckel syndromes
- PMID: 33791682
- PMCID: PMC8009330
- DOI: 10.1016/j.xhgg.2020.100016
TMEM218 dysfunction causes ciliopathies, including Joubert and Meckel syndromes
Abstract
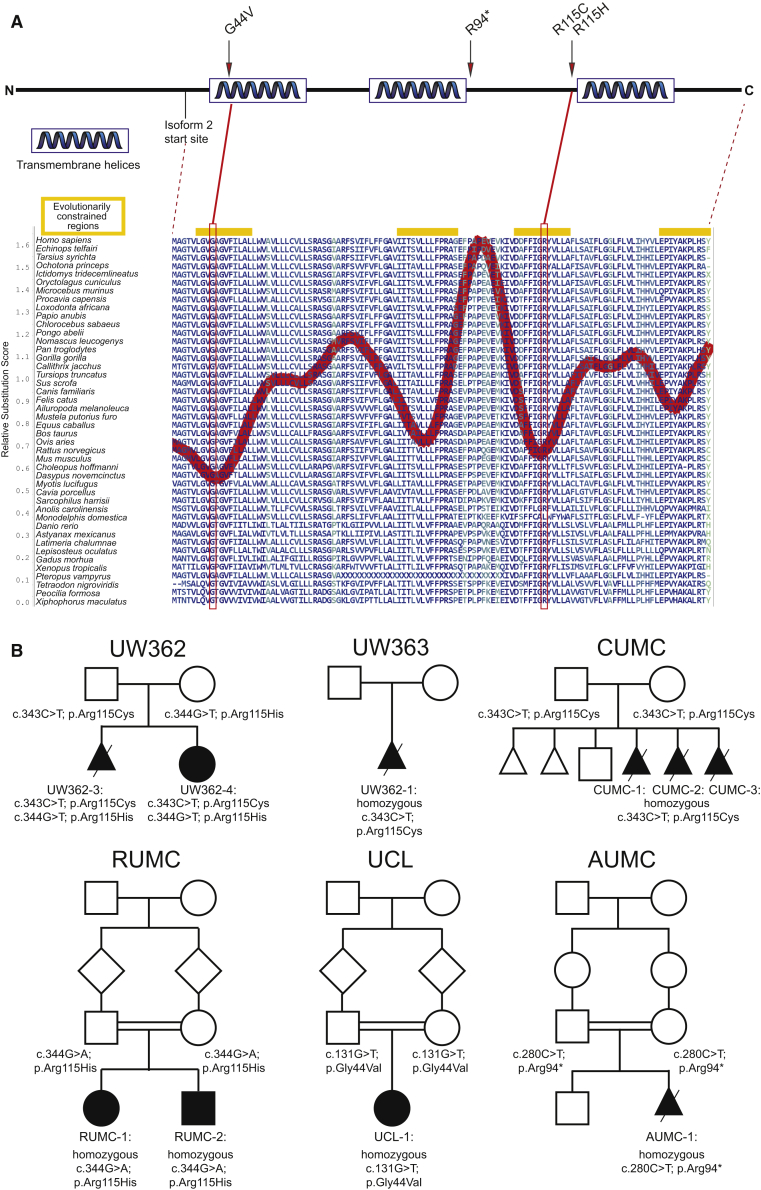
The Joubert-Meckel syndrome spectrum is a continuum of recessive ciliopathy conditions caused by primary cilium dysfunction. The primary cilium is a microtubule-based, antenna-like organelle that projects from the surface of most human cell types, allowing them to respond to extracellular signals. The cilium is partitioned from the cell body by the transition zone, a known hotspot for ciliopathy-related proteins. Despite years of Joubert syndrome (JBTS) gene discovery, the genetic cause cannot be identified in up to 30% of individuals with JBTS, depending on the cohort, sequencing method, and criteria for pathogenic variants. Using exome and targeted sequencing of 655 families with JBTS, we identified three individuals from two families harboring biallelic, rare, predicted-deleterious missense TMEM218 variants. Via MatchMaker Exchange, we identified biallelic TMEM218 variants in four additional families with ciliopathy phenotypes. Of note, four of the six families carry missense variants affecting the same highly conserved amino acid position 115. Clinical features included the molar tooth sign (N = 2), occipital encephalocele (N = 5, all fetuses), retinal dystrophy (N = 4, all living individuals), polycystic kidneys (N = 2), and polydactyly (N = 2), without liver involvement. Combined with existing functional data linking TMEM218 to ciliary transition zone function, our human genetic data make a strong case for TMEM218 dysfunction as a cause of ciliopathy phenotypes including JBTS with retinal dystrophy and Meckel syndrome. Identifying all genetic causes of the Joubert-Meckel spectrum enables diagnostic testing, prognostic and recurrence risk counseling, and medical monitoring, as well as work to delineate the underlying biological mechanisms and identify targets for future therapies.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures

References
-
- Joubert M., Eisenring J.J., Robb J.P., Andermann F. Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology. 1969;19:813–825. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
