

This is a preprint.
Freely accessible ready to use global infrastructure for SARS-CoV-2 monitoring
- PMID: 33791701
- PMCID: PMC8010728
- DOI: 10.1101/2021.03.25.437046
Freely accessible ready to use global infrastructure for SARS-CoV-2 monitoring
Update in
-
Ready-to-use public infrastructure for global SARS-CoV-2 monitoring.Nat Biotechnol. 2021 Oct;39(10):1178-1179. doi: 10.1038/s41587-021-01069-1. Nat Biotechnol. 2021. PMID: 34588690 Free PMC article. No abstract available.
Abstract
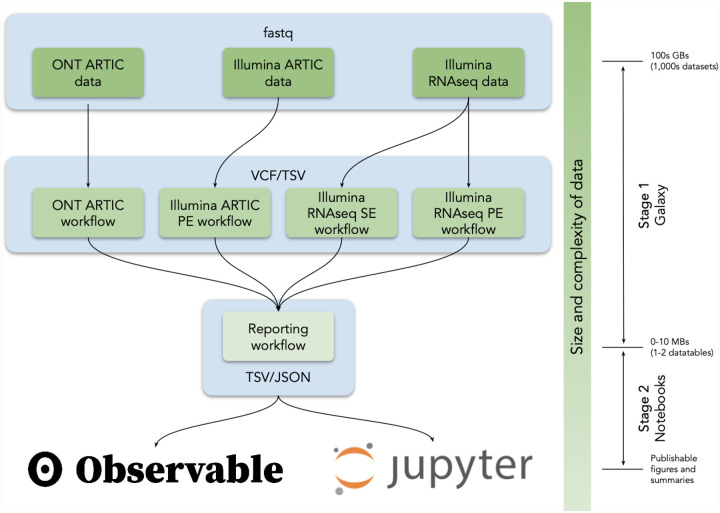
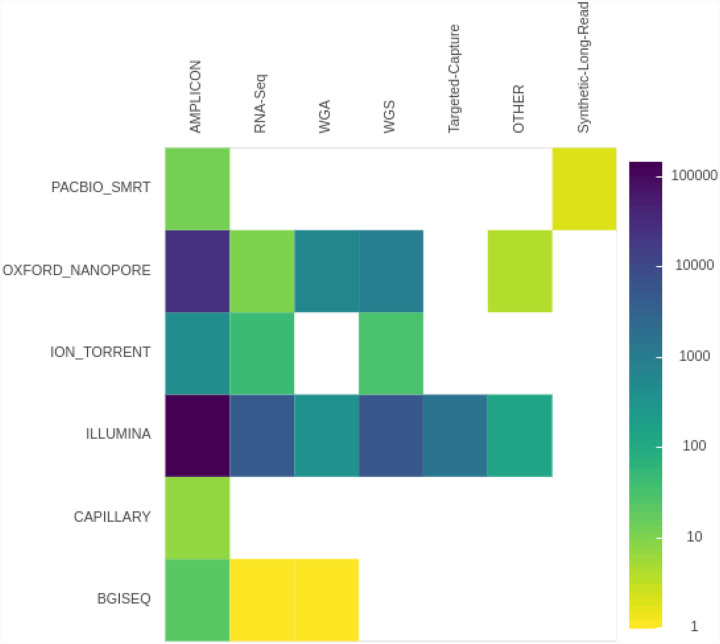
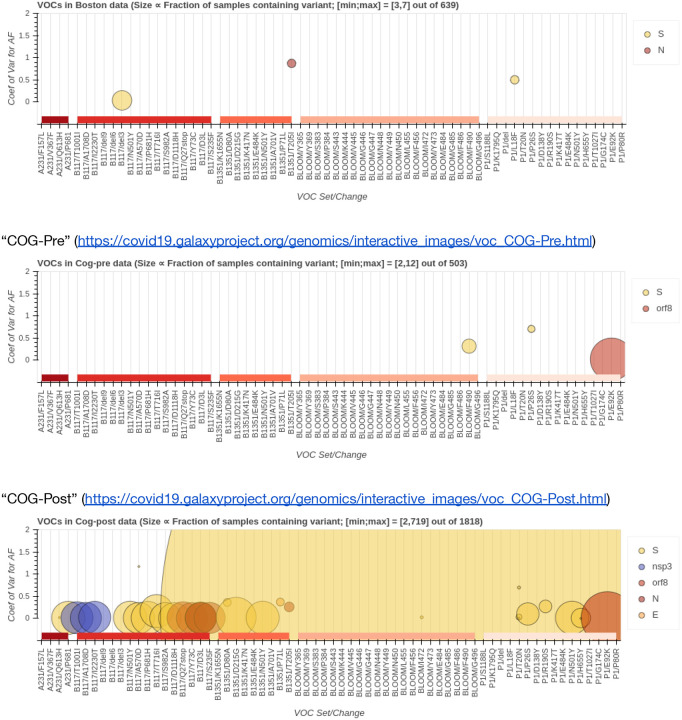
The COVID-19 pandemic is the first global health crisis to occur in the age of big genomic data.Although data generation capacity is well established and sufficiently standardized, analytical capacity is not. To establish analytical capacity it is necessary to pull together global computational resources and deliver the best open source tools and analysis workflows within a ready to use, universally accessible resource. Such a resource should not be controlled by a single research group, institution, or country. Instead it should be maintained by a community of users and developers who ensure that the system remains operational and populated with current tools. A community is also essential for facilitating the types of discourse needed to establish best analytical practices. Bringing together public computational research infrastructure from the USA, Europe, and Australia, we developed a distributed data analysis platform that accomplishes these goals. It is immediately accessible to anyone in the world and is designed for the analysis of rapidly growing collections of deep sequencing datasets. We demonstrate its utility by detecting allelic variants in high-quality existing SARS-CoV-2 sequencing datasets and by continuous reanalysis of COG-UK data. All workflows, data, and documentation is available at https://covid19.galaxyproject.org .
Figures

References
-
- XSEDE. www.xsede.org.
-
- ELIXIR-DE. https://www.denbi.de/elixir-de.
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous