

Targeted replacement of full-length CFTR in human airway stem cells by CRISPR-Cas9 for pan-mutation correction in the endogenous locus
- PMID: 33794364
- PMCID: PMC8753290
- DOI: 10.1016/j.ymthe.2021.03.023
Targeted replacement of full-length CFTR in human airway stem cells by CRISPR-Cas9 for pan-mutation correction in the endogenous locus
Abstract
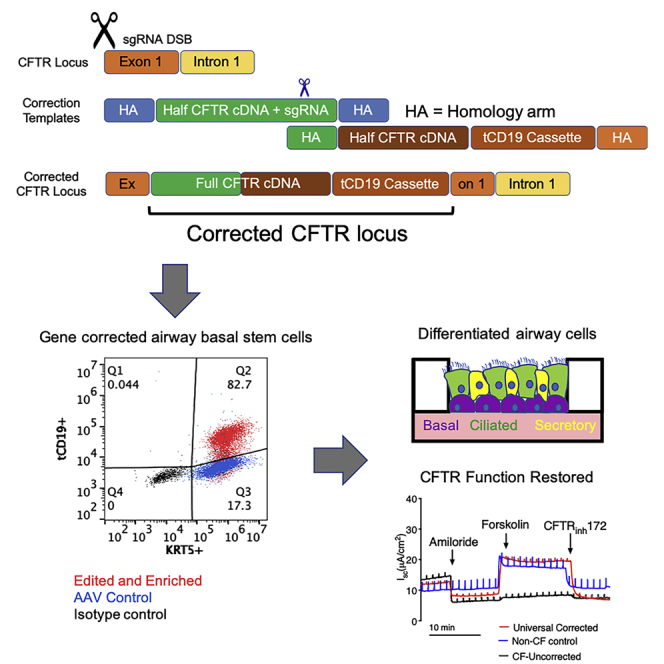
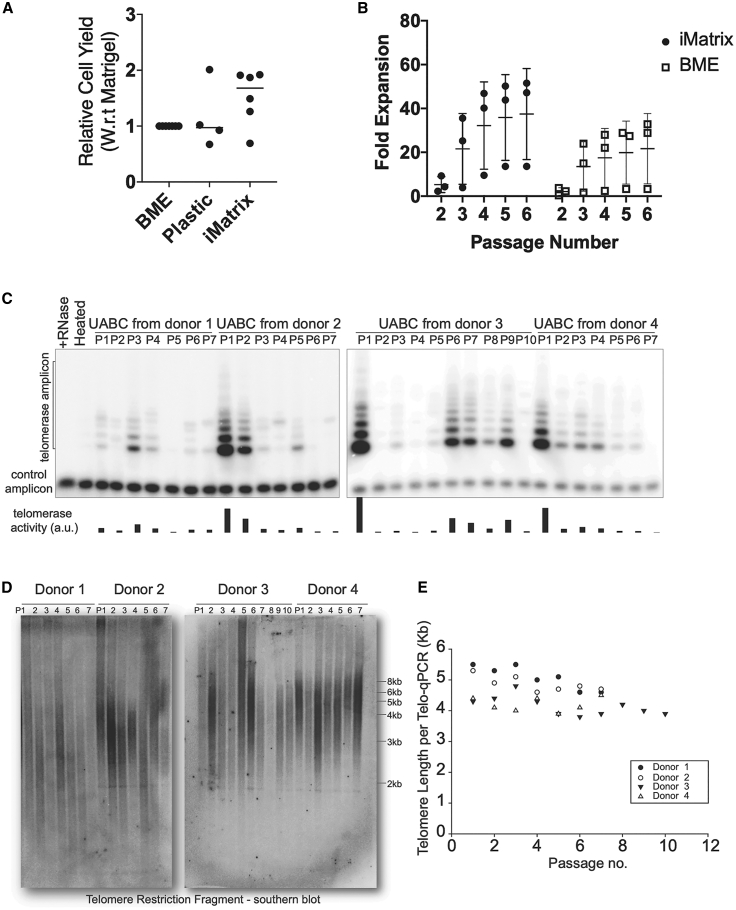
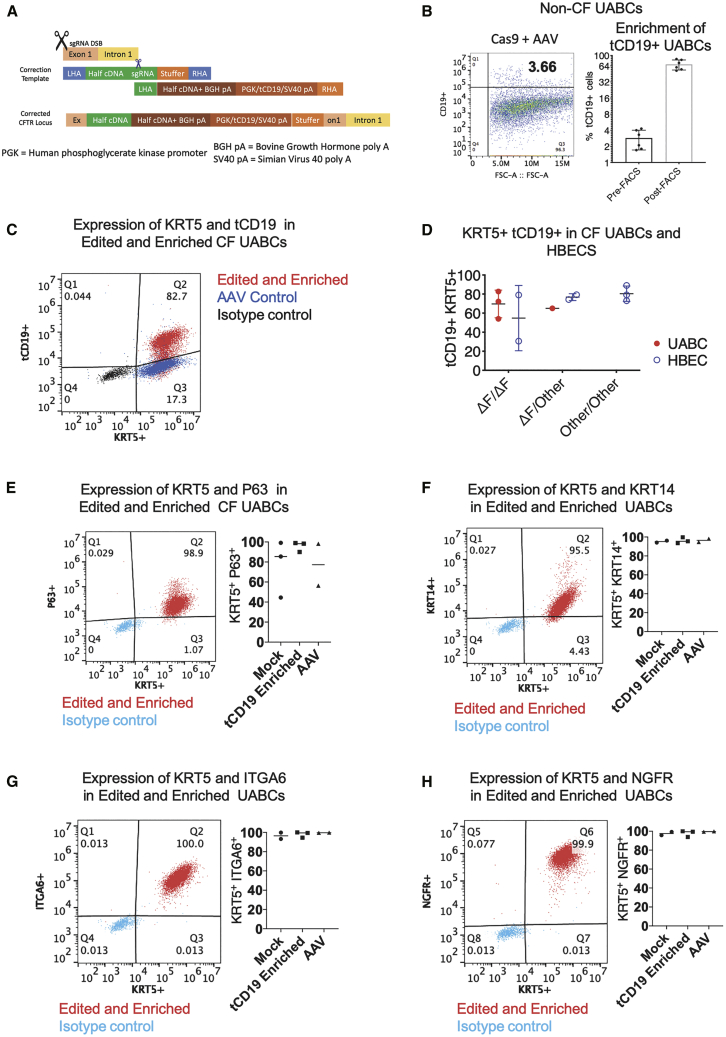
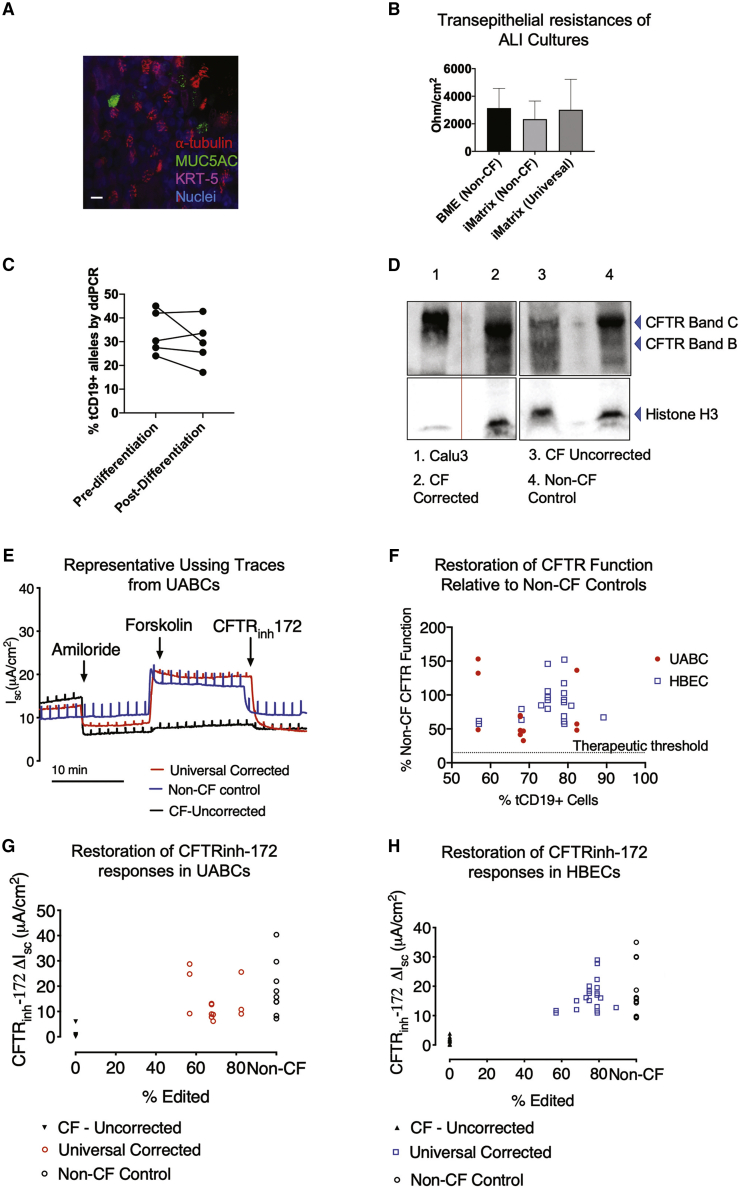
Cystic fibrosis (CF) is a monogenic disease caused by impaired production and/or function of the CF transmembrane conductance regulator (CFTR) protein. Although we have previously shown correction of the most common pathogenic mutation, there are many other pathogenic mutations throughout the CF gene. An autologous airway stem cell therapy in which the CFTR cDNA is precisely inserted into the CFTR locus may enable the development of a durable cure for almost all CF patients, irrespective of the causal mutation. Here, we use CRISPR-Cas9 and two adeno-associated viruses (AAVs) carrying the two halves of the CFTR cDNA to sequentially insert the full CFTR cDNA along with a truncated CD19 (tCD19) enrichment tag in upper airway basal stem cells (UABCs) and human bronchial epithelial cells (HBECs). The modified cells were enriched to obtain 60%-80% tCD19+ UABCs and HBECs from 11 different CF donors with a variety of mutations. Differentiated epithelial monolayers cultured at air-liquid interface showed restored CFTR function that was >70% of the CFTR function in non-CF controls. Thus, our study enables the development of a therapy for almost all CF patients, including patients who cannot be treated using recently approved modulator therapies.
Keywords: CF; CFTR correction; CRISPR-Cas9; airway stem cell therapy; cystic fibrosis; genome editing for CF; universal CFTR correction.
Copyright © 2021 The American Society of Gene and Cell Therapy. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests M.H.P. has equity and serves on the Scientific Advisory Board of CRISPR Therapeutics and Graphite Bio. J.V.N. is a consultant with COOK Medical, which manufactures the pSIS graft. Neither company had any input on the design, execution, interpretation, or publication of the work in this manuscript.
Figures
References
-
- Taylor-Cousar J.L., Munck A., McKone E.F., van der Ent C.K., Moeller A., Simard C., Wang L.T., Ingenito E.P., McKee C., Lu Y., et al. Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N. Engl. J. Med. 2017;377:2013–2023. - PubMed
-
- Heijerman H.G.M., McKone E.F., Downey D.G., Van Braeckel E., Rowe S.M., Tullis E., Mall M.A., Welter J.J., Ramsey B.W., McKee C.M., et al. VX17-445-103 Trial Group Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. Lancet. 2019;394:1940–1948. - PMC - PubMed
-
- Konstan M.W., Flume P.A. Clinical care for cystic fibrosis: preparing for the future now. Lancet Respir. Med. 2020;8:10–12. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- K08 DK124684/DK/NIDDK NIH HHS/United States
- U01 CA176299/CA/NCI NIH HHS/United States
- R01 HL151677/HL/NHLBI NIH HHS/United States
- U24 DK085532/DK/NIDDK NIH HHS/United States
- U01 DK085532/DK/NIDDK NIH HHS/United States
- R35 CA197563/CA/NCI NIH HHS/United States
- R01 AG056575/AG/NIA NIH HHS/United States
- P30 CA124435/CA/NCI NIH HHS/United States
- P30 DK065988/DK/NIDDK NIH HHS/United States
- K08 DE027730/DE/NIDCR NIH HHS/United States
- U01 DK085527/DK/NIDDK NIH HHS/United States
- U01 CA217851/CA/NCI NIH HHS/United States
- U19 AI116484/AI/NIAID NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
