

Single-cell chromatin accessibility identifies pancreatic islet cell type- and state-specific regulatory programs of diabetes risk
- PMID: 33795864
- PMCID: PMC9037575
- DOI: 10.1038/s41588-021-00823-0
Single-cell chromatin accessibility identifies pancreatic islet cell type- and state-specific regulatory programs of diabetes risk
Abstract
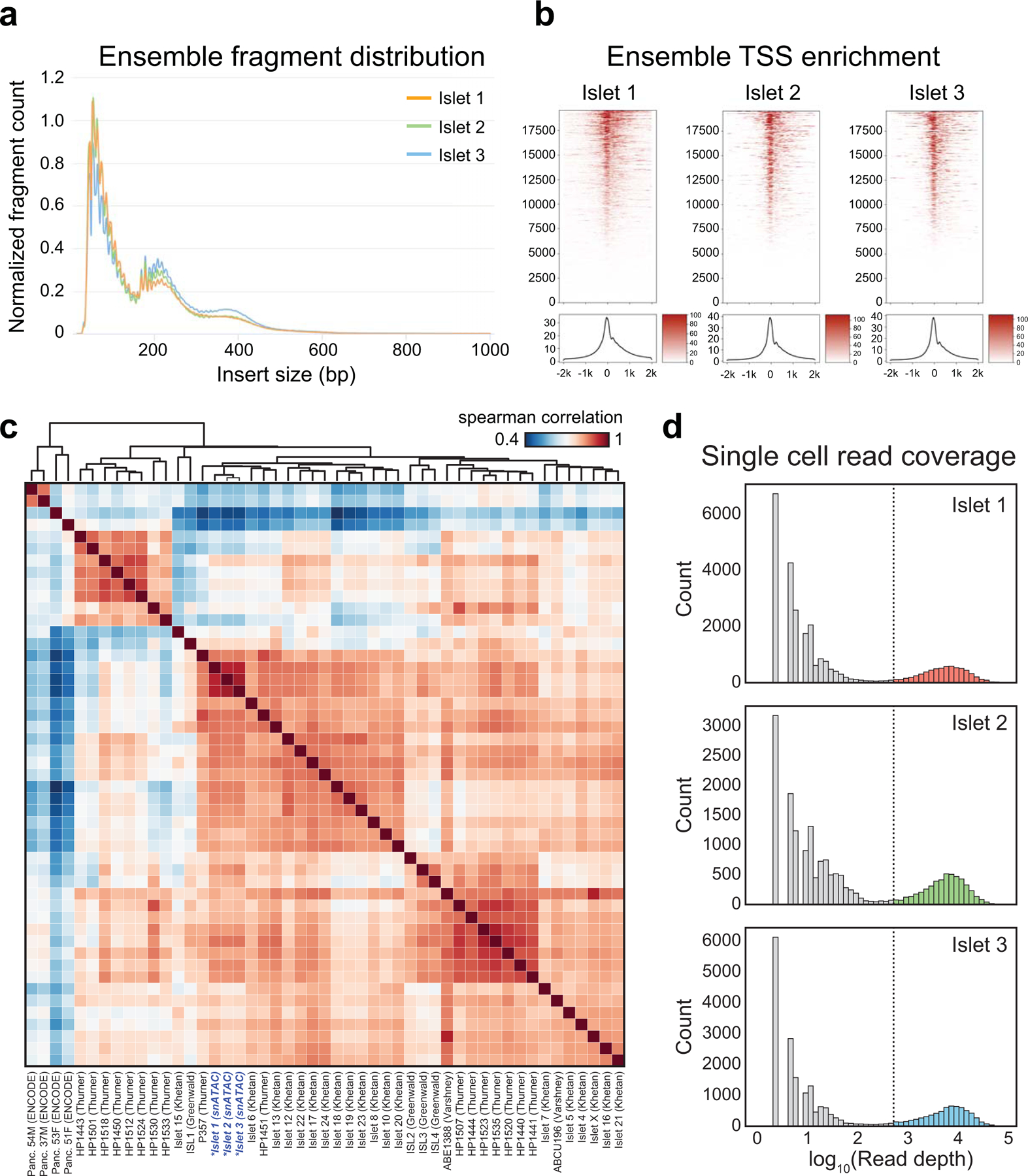
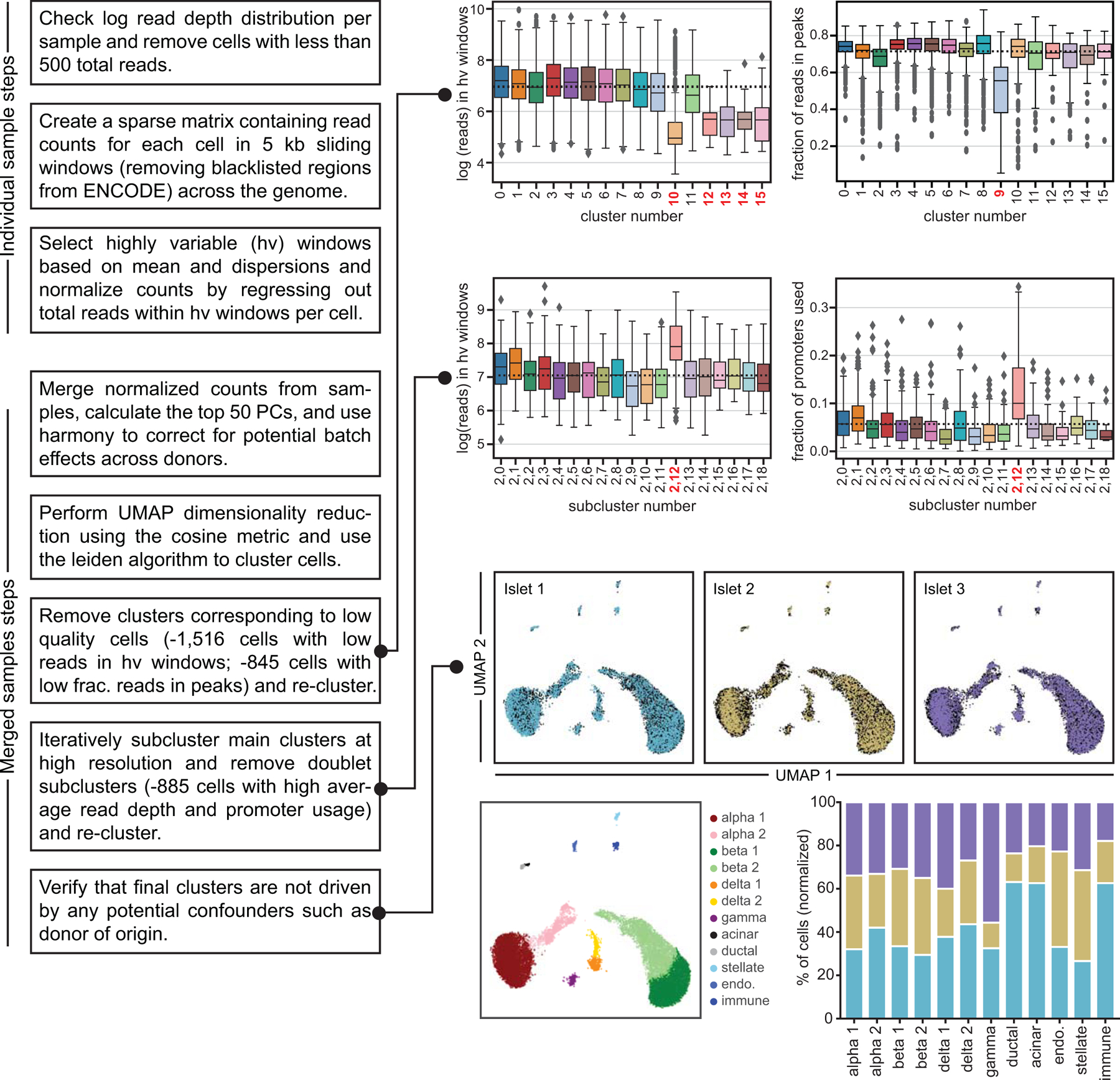
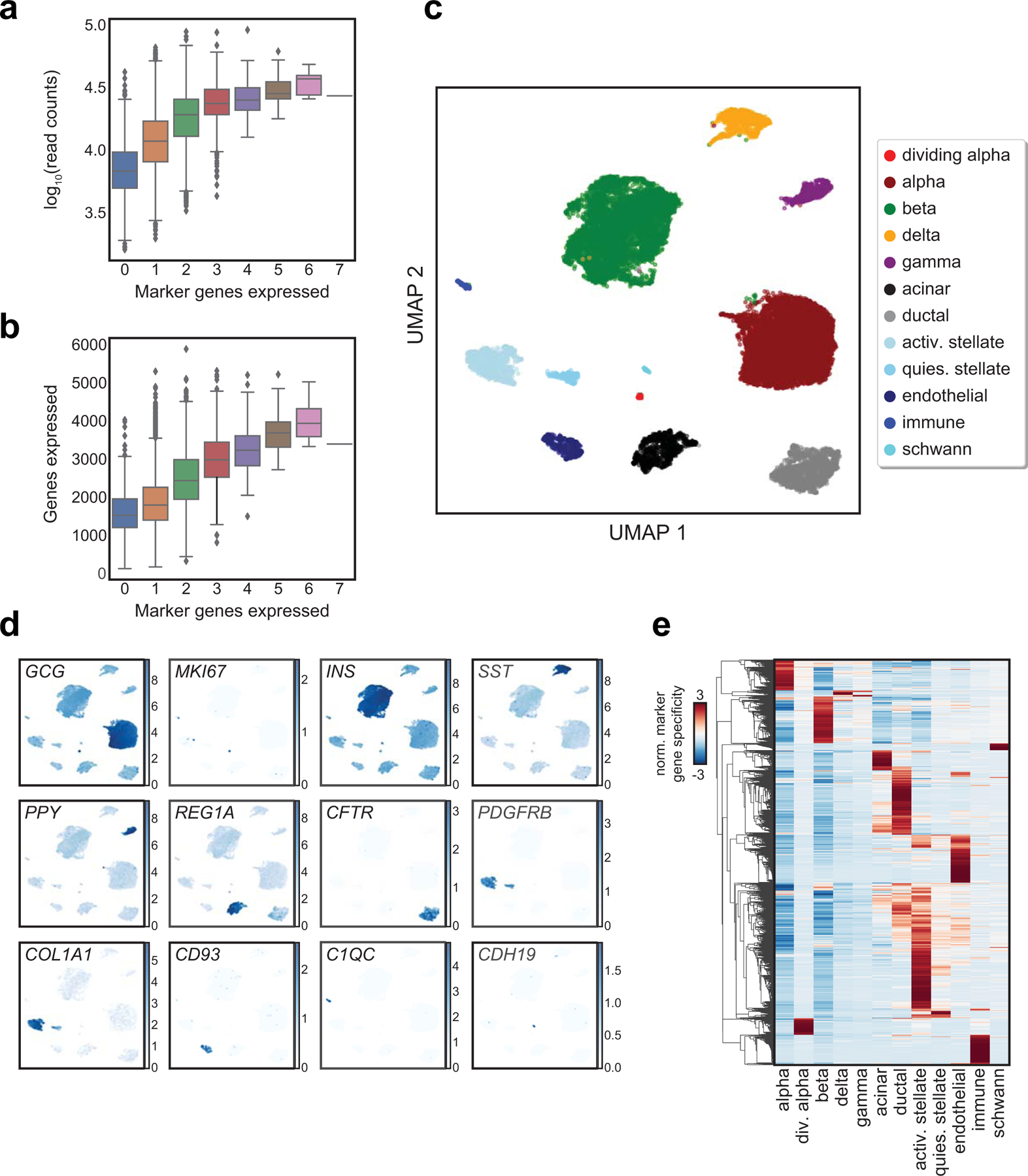
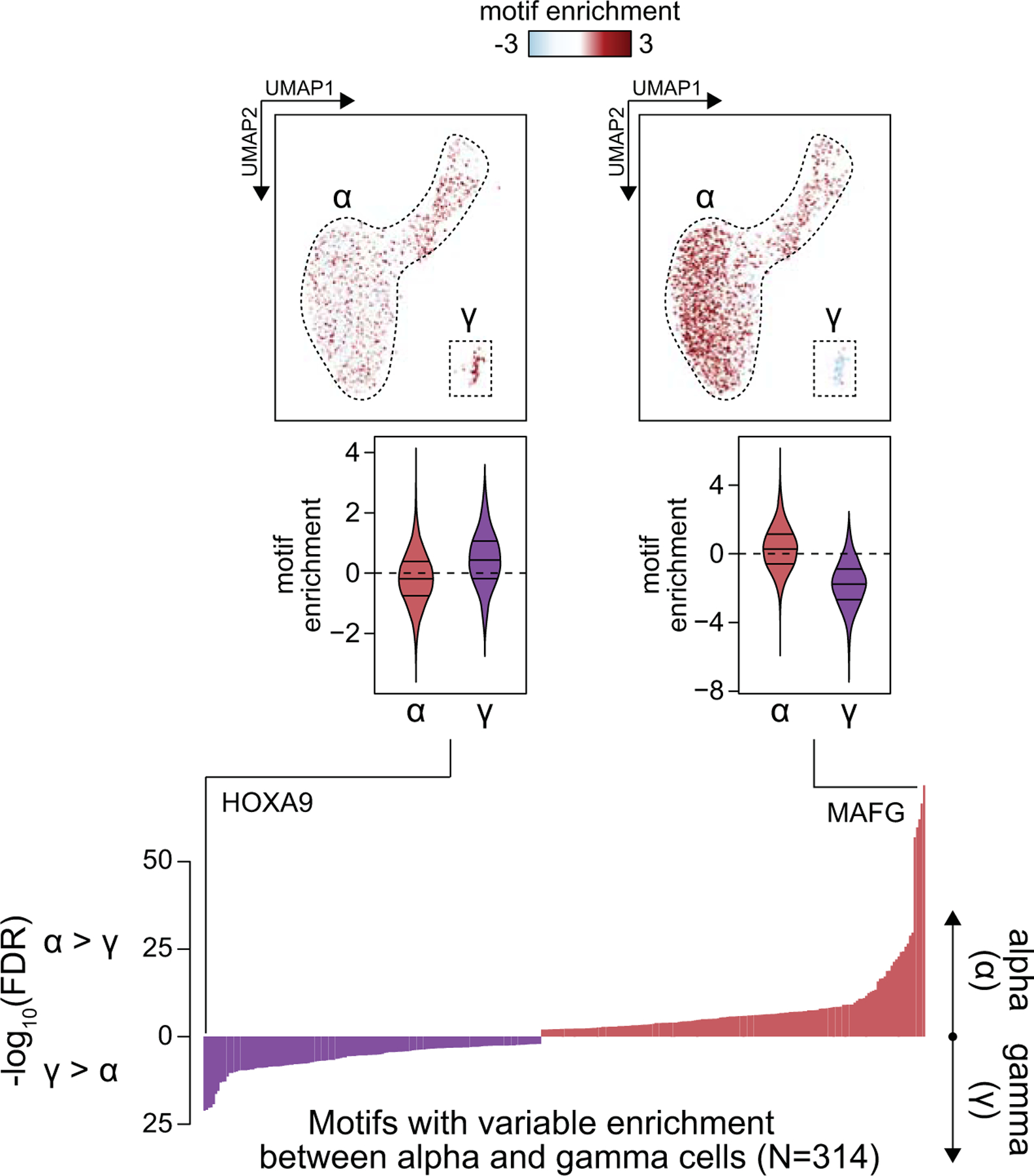
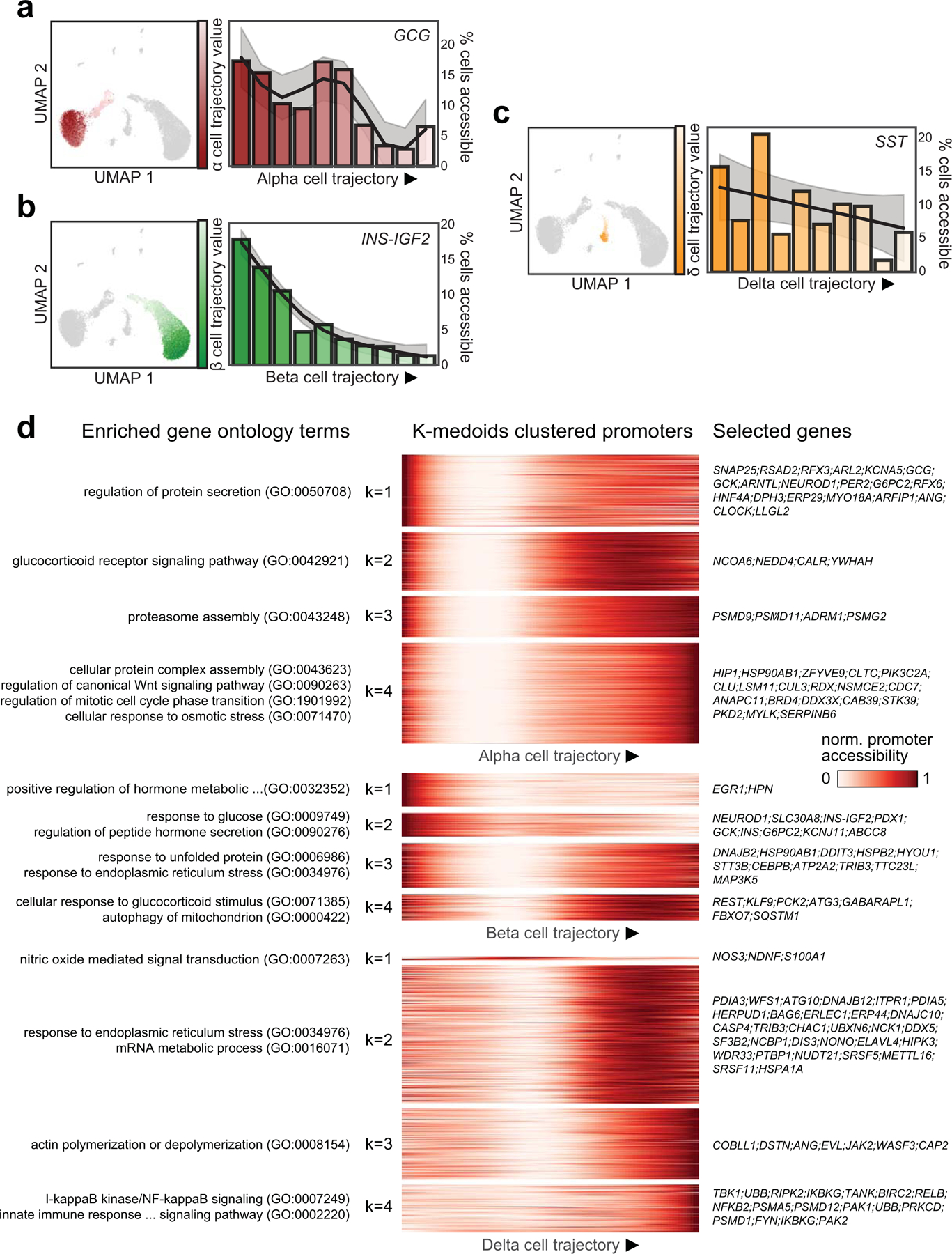
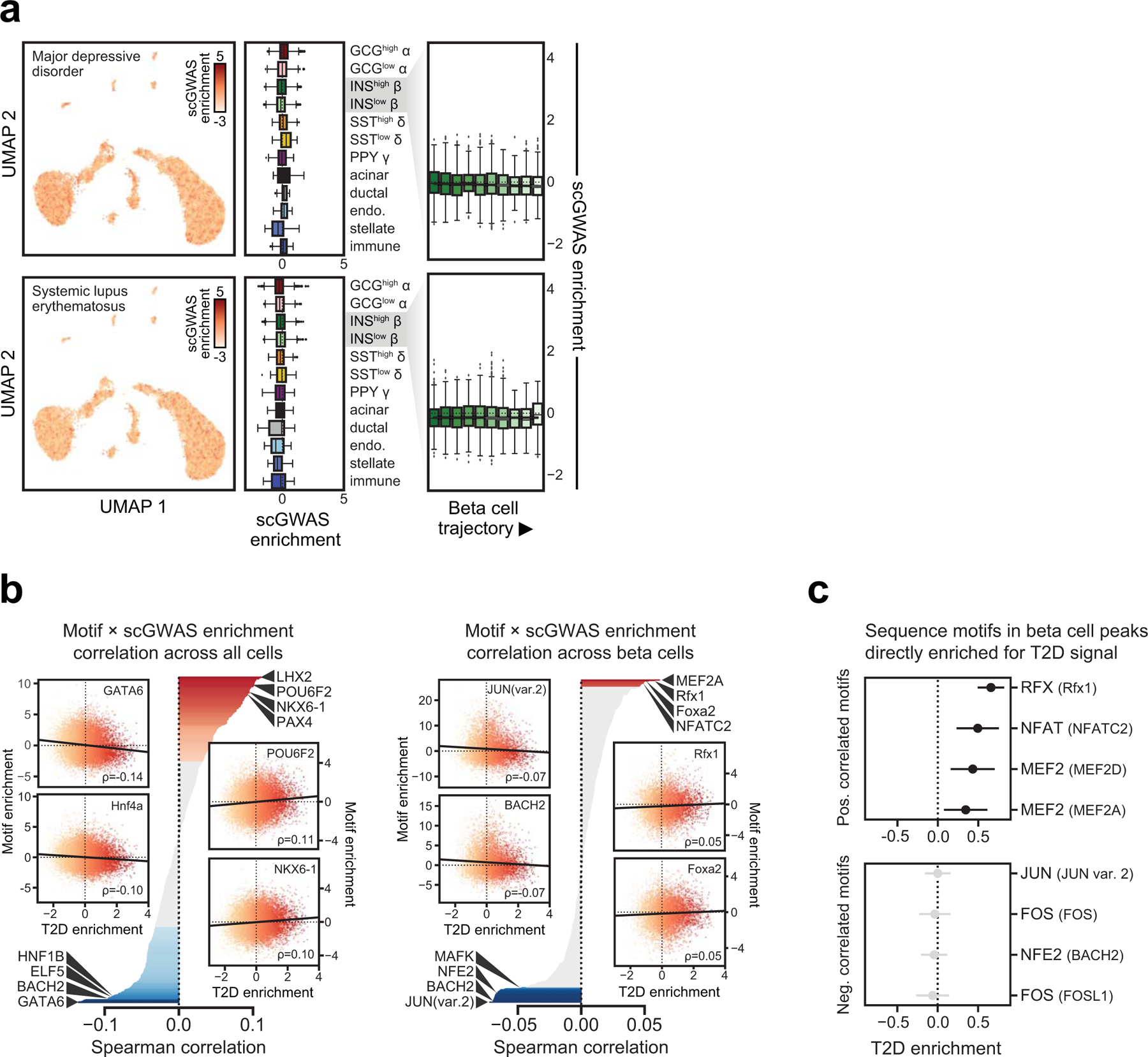
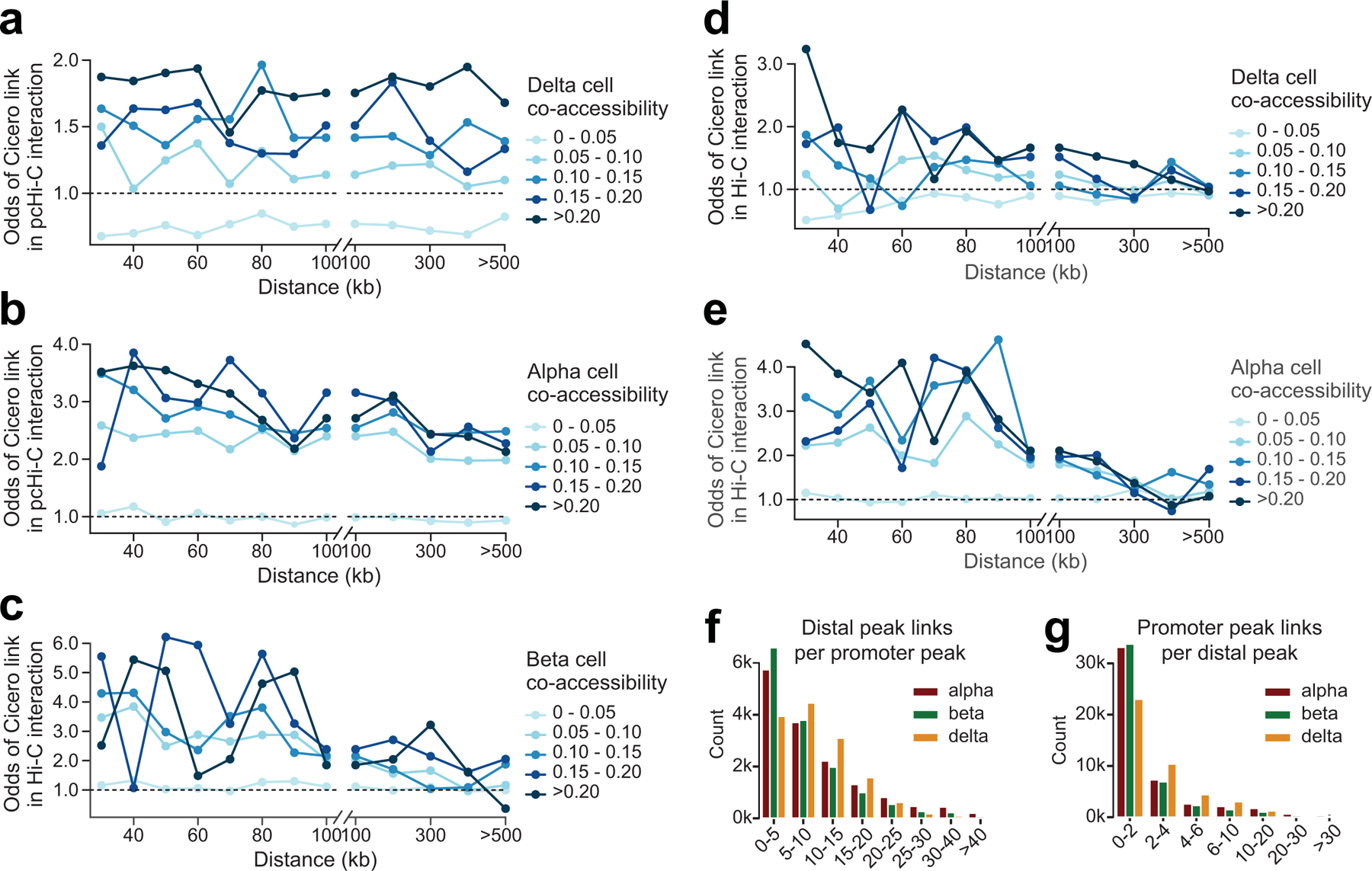
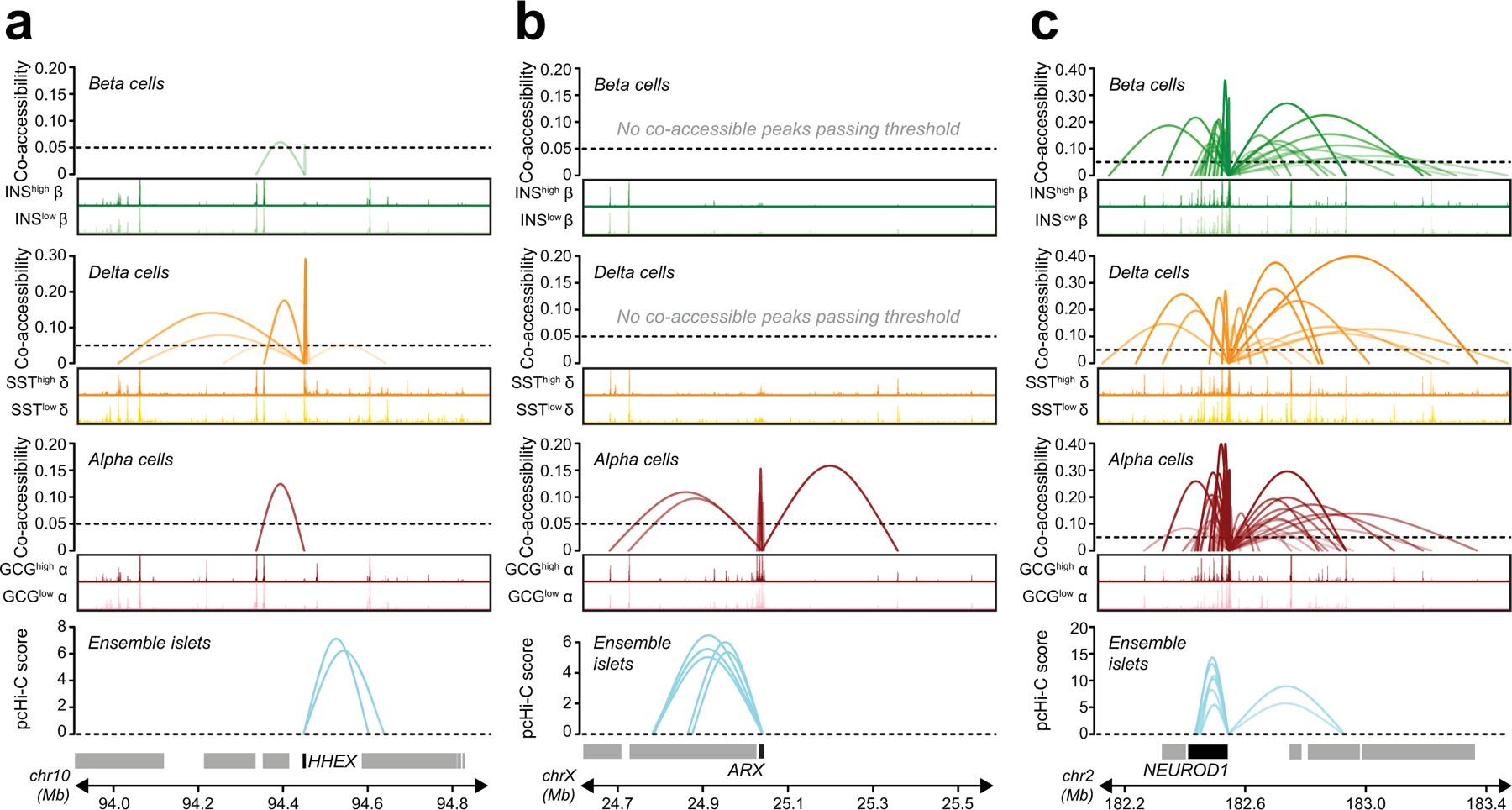
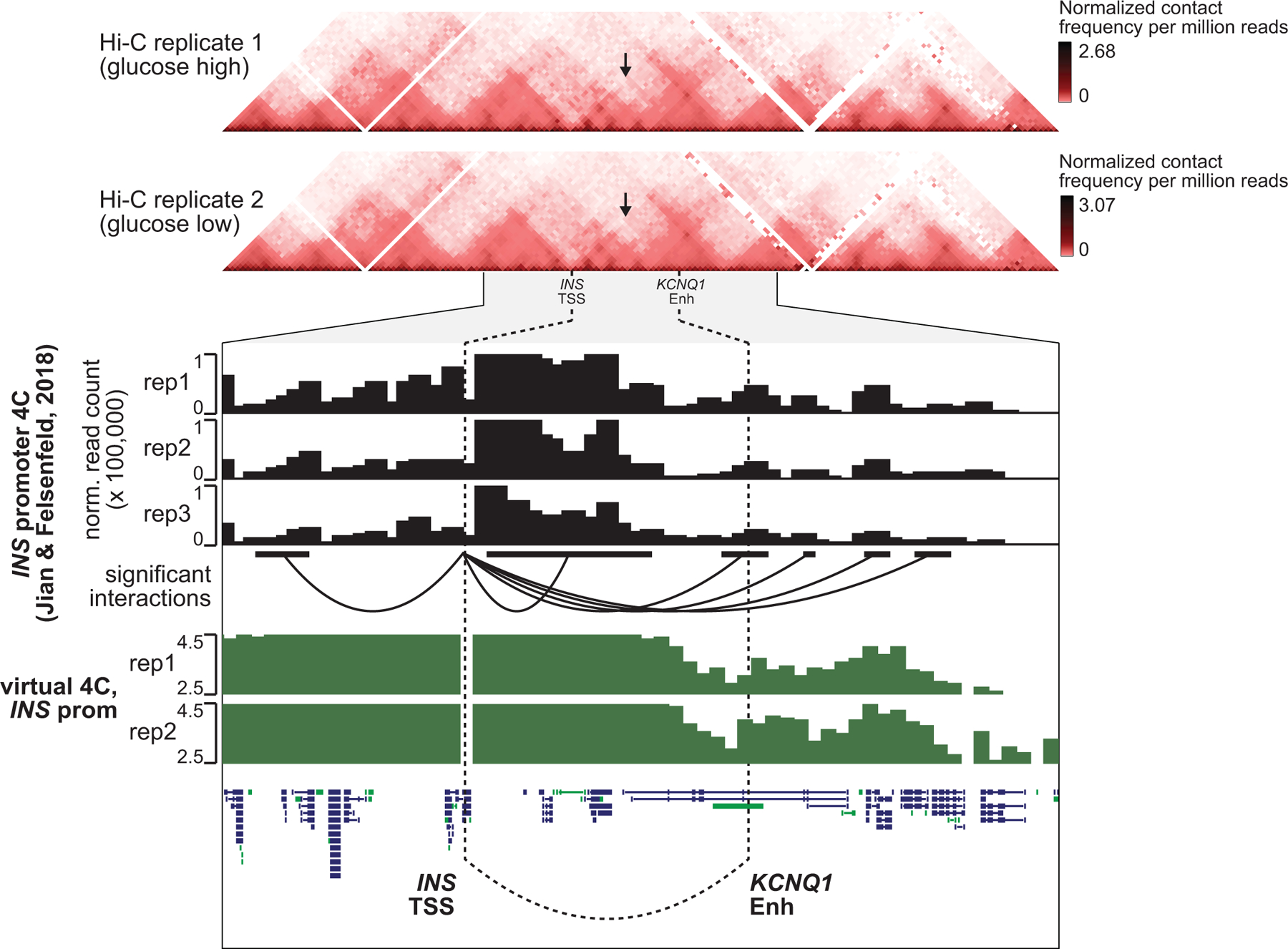
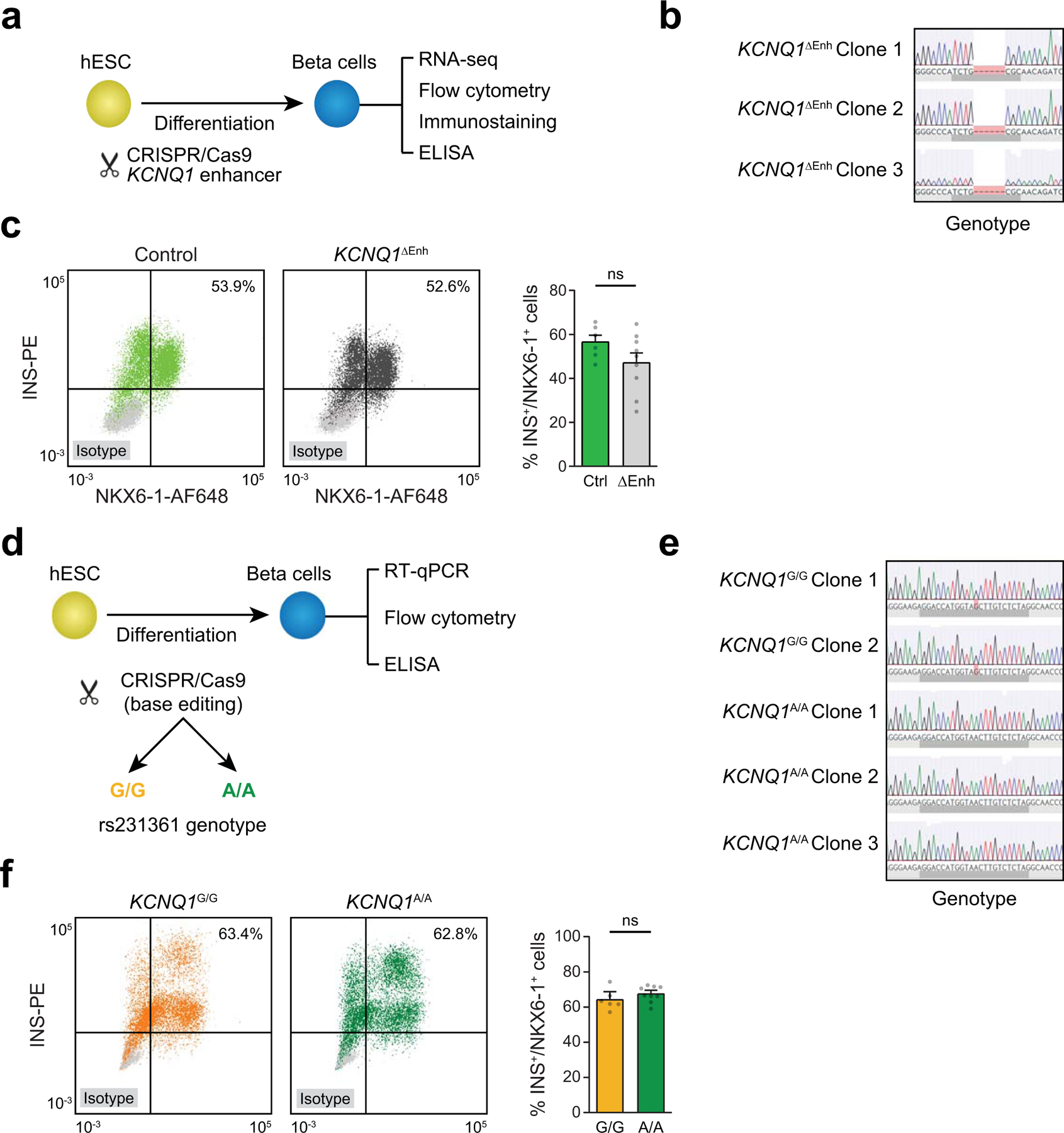
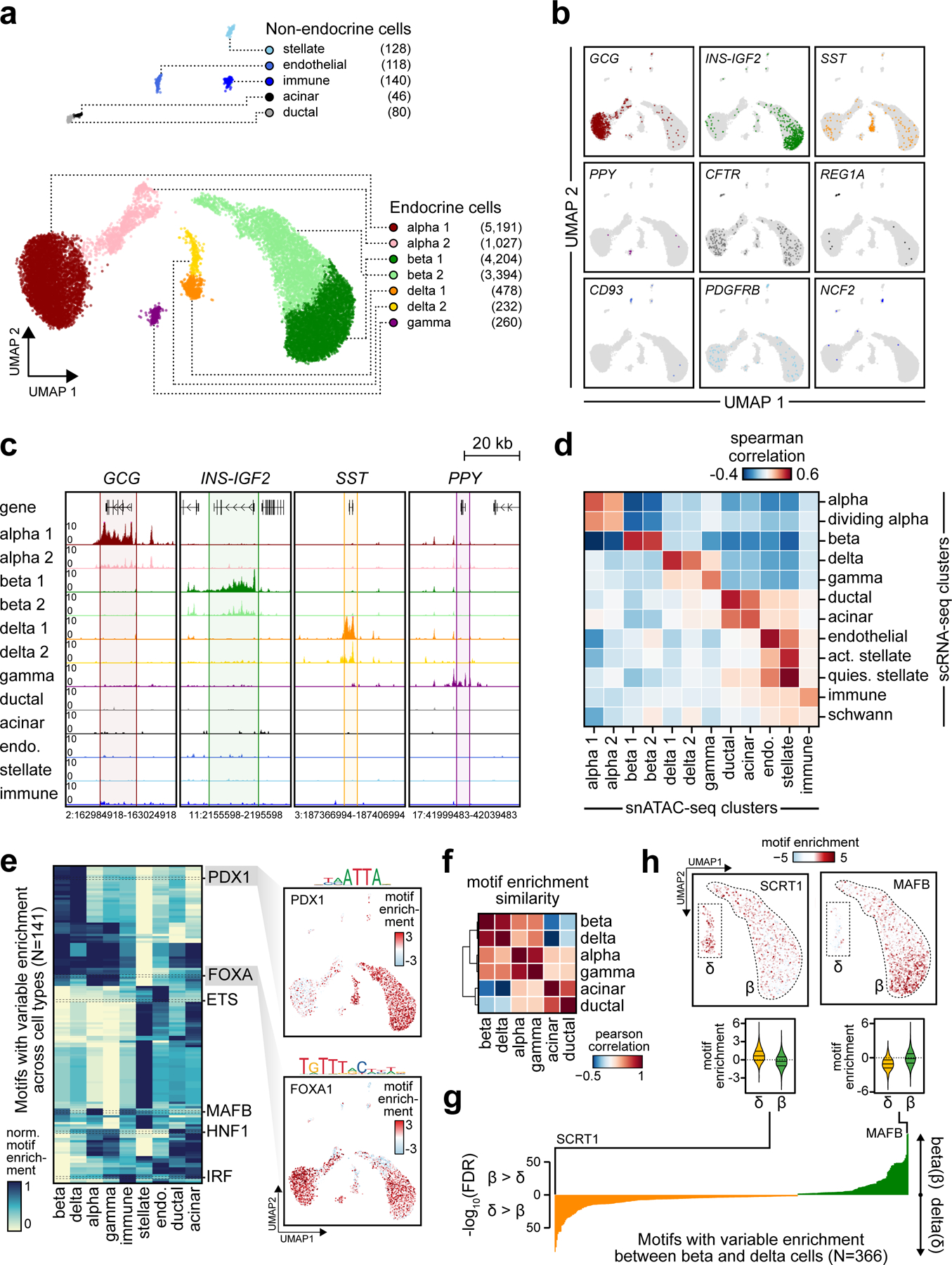
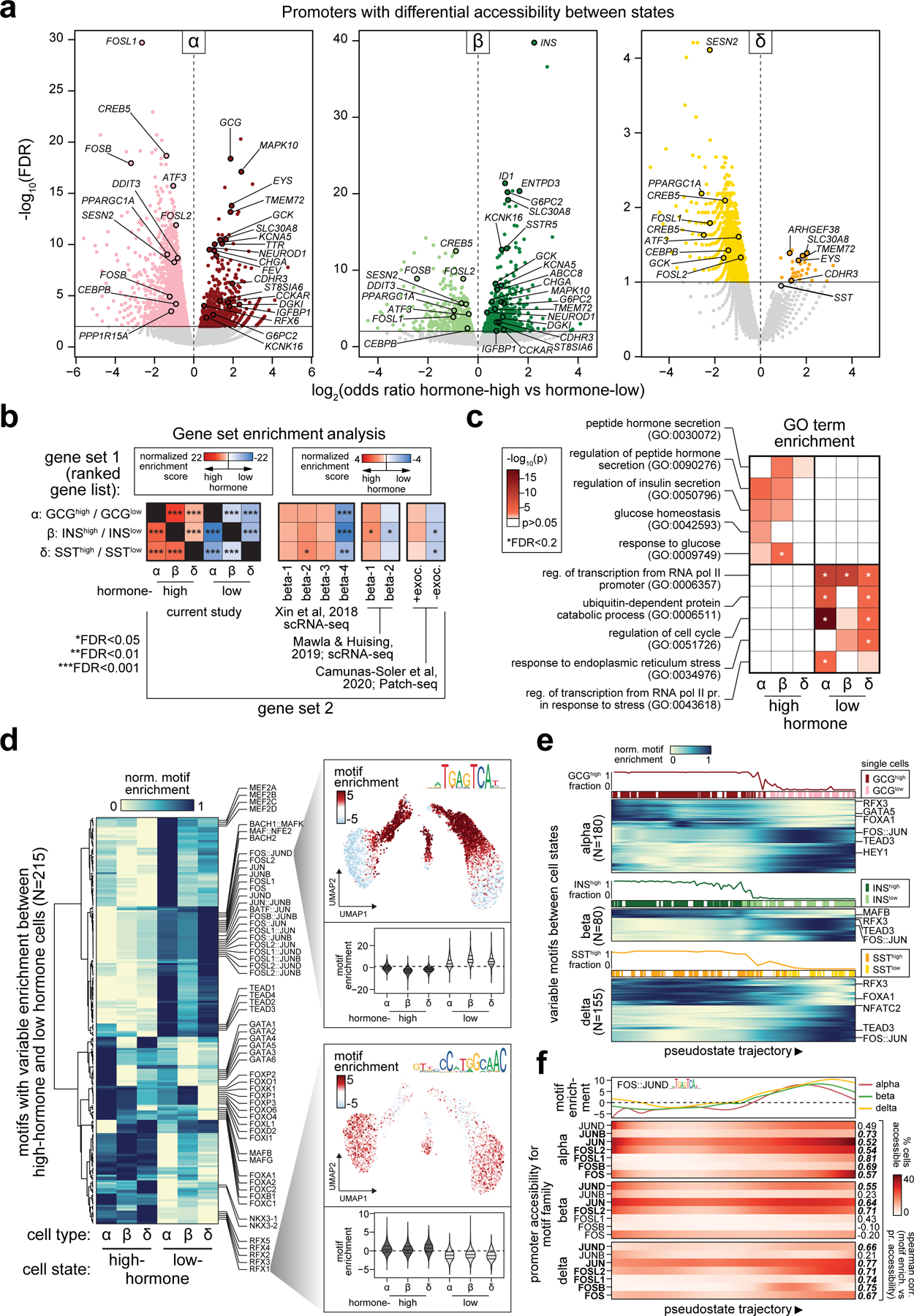
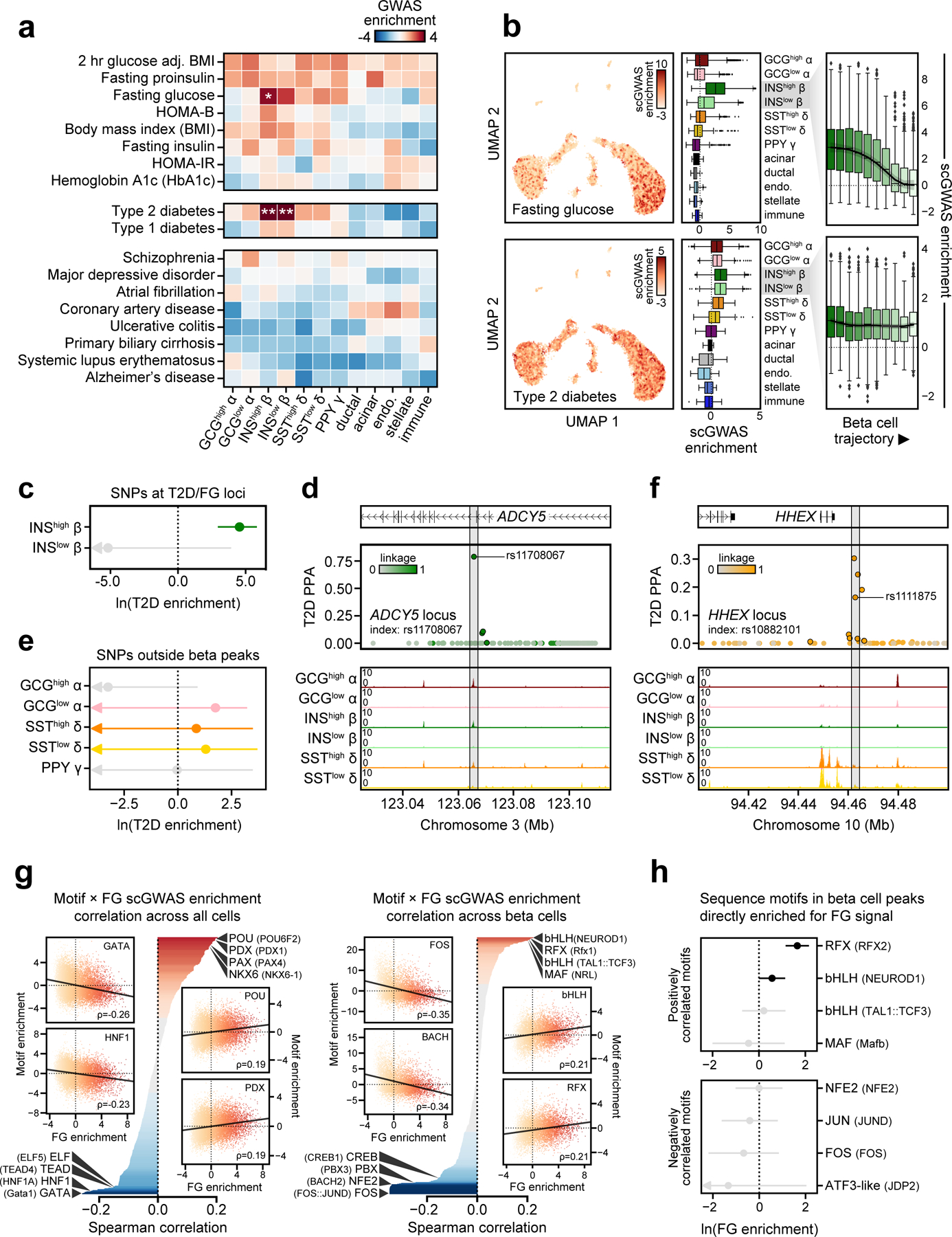
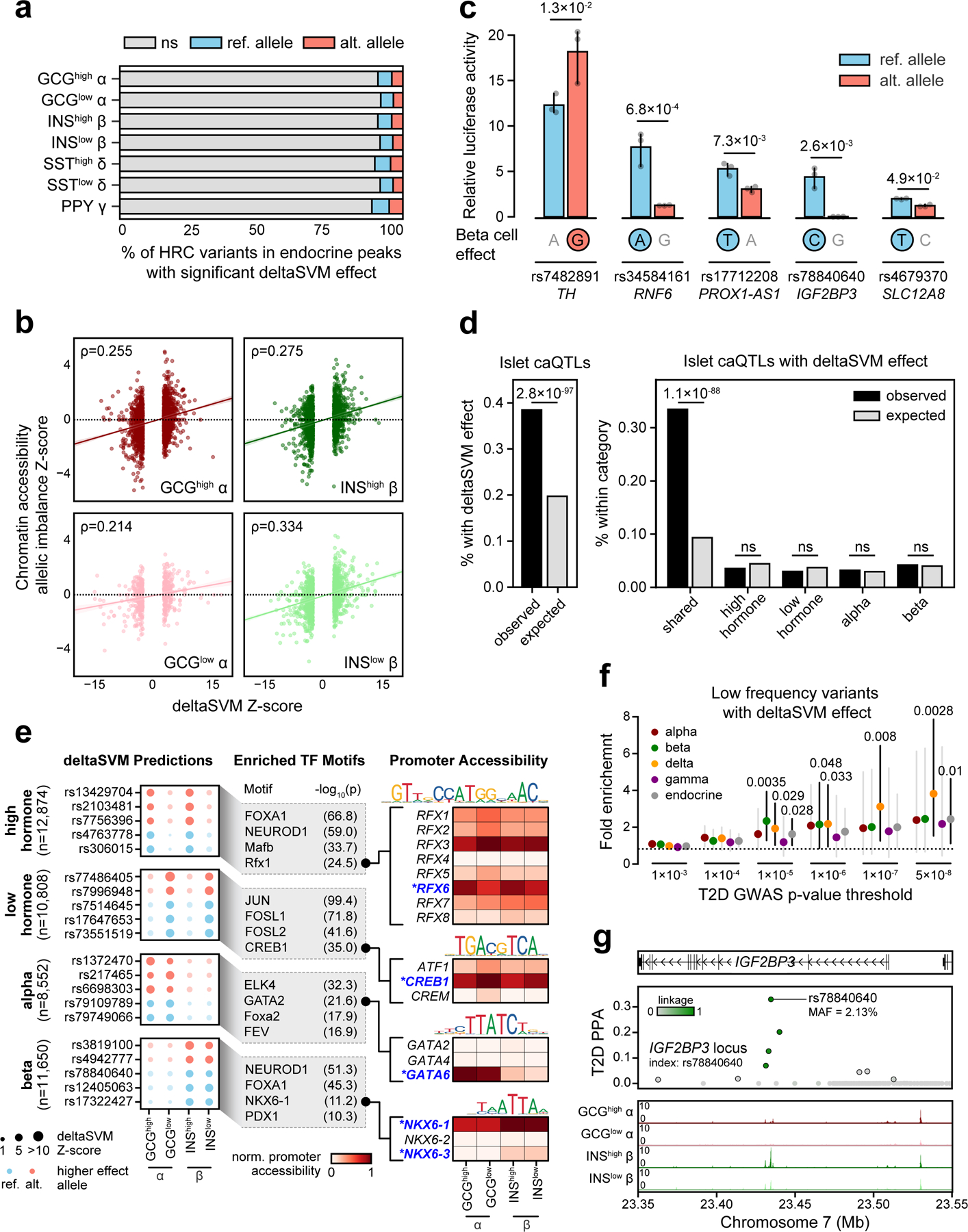
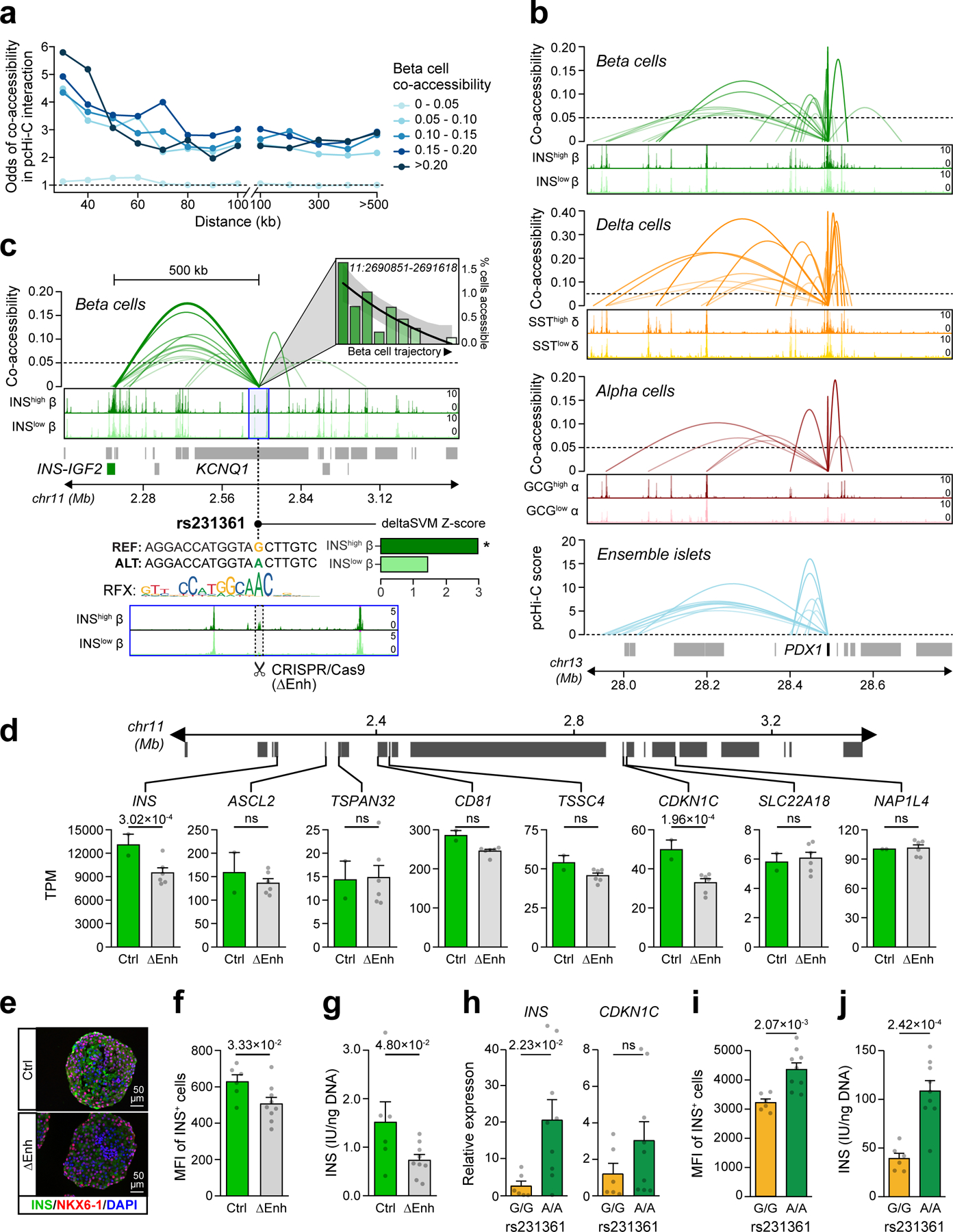
Single-nucleus assay for transposase-accessible chromatin using sequencing (snATAC-seq) creates new opportunities to dissect cell type-specific mechanisms of complex diseases. Since pancreatic islets are central to type 2 diabetes (T2D), we profiled 15,298 islet cells by using combinatorial barcoding snATAC-seq and identified 12 clusters, including multiple alpha, beta and delta cell states. We cataloged 228,873 accessible chromatin sites and identified transcription factors underlying lineage- and state-specific regulation. We observed state-specific enrichment of fasting glucose and T2D genome-wide association studies for beta cells and enrichment for other endocrine cell types. At T2D signals localized to islet-accessible chromatin, we prioritized variants with predicted regulatory function and co-accessibility with target genes. A causal T2D variant rs231361 at the KCNQ1 locus had predicted effects on a beta cell enhancer co-accessible with INS and genome editing in embryonic stem cell-derived beta cells affected INS levels. Together our findings demonstrate the power of single-cell epigenomics for interpreting complex disease genetics.
Conflict of interest statement
COMPETING INTERESTS STATEMENT
K.J.G. does consulting for Genentech and holds stock in Vertex Pharmaceuticals, neither of which is related to the work in this study. No other authors have competing interests to disclose.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
