

Iron Metabolism Disorders for Cognitive Dysfunction After Mild Traumatic Brain Injury
- PMID: 33796002
- PMCID: PMC8007909
- DOI: 10.3389/fnins.2021.587197
Iron Metabolism Disorders for Cognitive Dysfunction After Mild Traumatic Brain Injury
Abstract
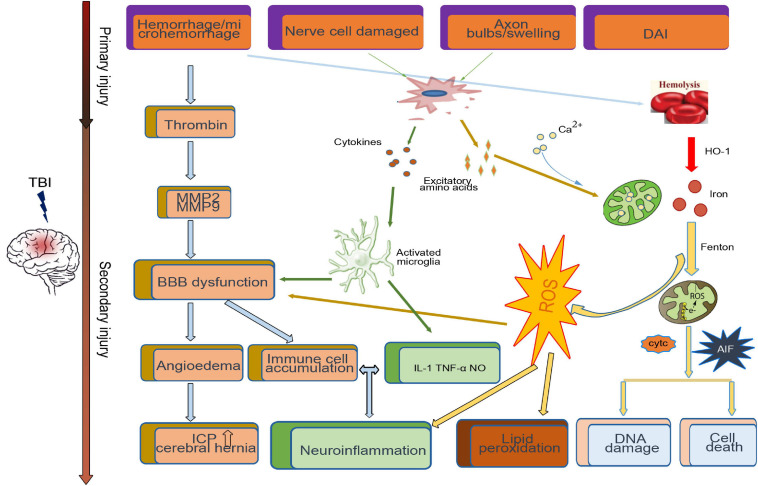
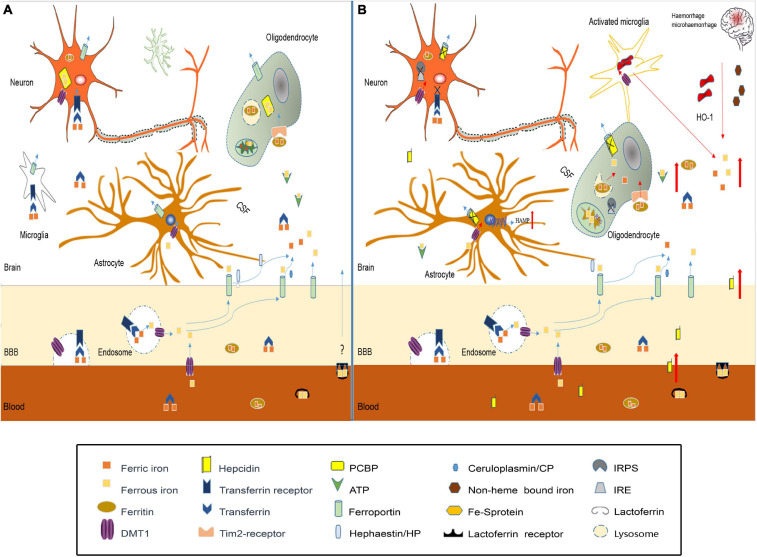
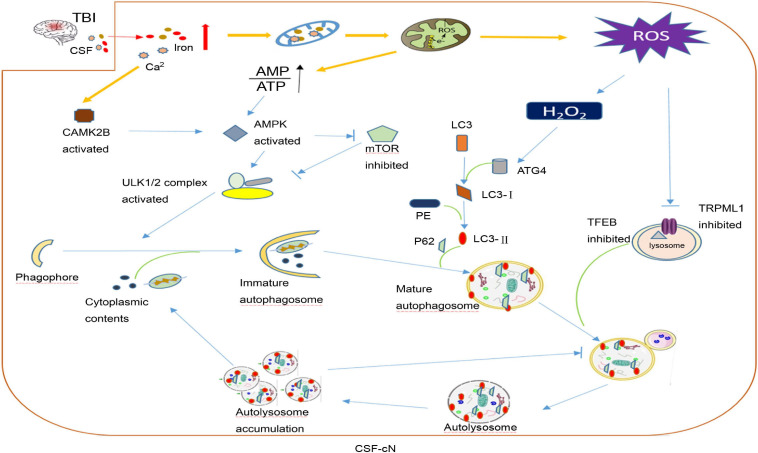
Traumatic brain injury (TBI) is one of the most harmful forms of acute brain injury and predicted to be one of the three major neurological diseases that cause neurological disabilities by 2030. A series of secondary injury cascades often cause cognitive dysfunction of TBI patients leading to poor prognosis. However, there are still no effective intervention measures, which drive us to explore new therapeutic targets. In this process, the most part of mild traumatic brain injury (mTBI) is ignored because its initial symptoms seemed not serious. Unfortunately, the ignored mTBI accounts for 80% of the total TBI, and a large part of the patients have long-term cognitive dysfunction. Iron deposition has been observed in mTBI patients and accompanies the whole pathological process. Iron accumulation may affect long-term cognitive dysfunction from three pathways: local injury, iron deposition induces tau phosphorylation, the formation of neurofibrillary tangles; neural cells death; and neural network damage, iron deposition leads to axonal injury by utilizing the iron sensibility of oligodendrocytes. Thus, iron overload and metabolism dysfunction was thought to play a pivotal role in mTBI pathophysiology. Cerebrospinal fluid-contacting neurons (CSF-cNs) located in the ependyma have bidirectional communication function between cerebral-spinal fluid and brain parenchyma, and may participate in the pathway of iron-induced cognitive dysfunction through projected nerve fibers and transmitted factor, such as 5-hydroxytryptamine, etc. The present review provides an overview of the metabolism and function of iron in mTBI, and to seek a potential new treatment target for mTBI with a novel perspective through combined iron and CSF-cNs.
Keywords: autophagy; cerebrospinal-fluid contacting neuron; cognitive dysfunction; iron metabolism; traumatic brain injury.
Copyright © 2021 Huang, Li, Feng and Chen.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources