

A novel homozygous SLC13A5 whole-gene deletion generated by Alu/Alu-mediated rearrangement in an Iraqi family with epileptic encephalopathy
- PMID: 33797191
- PMCID: PMC8445493
- DOI: 10.1002/ajmg.a.62192
A novel homozygous SLC13A5 whole-gene deletion generated by Alu/Alu-mediated rearrangement in an Iraqi family with epileptic encephalopathy
Abstract
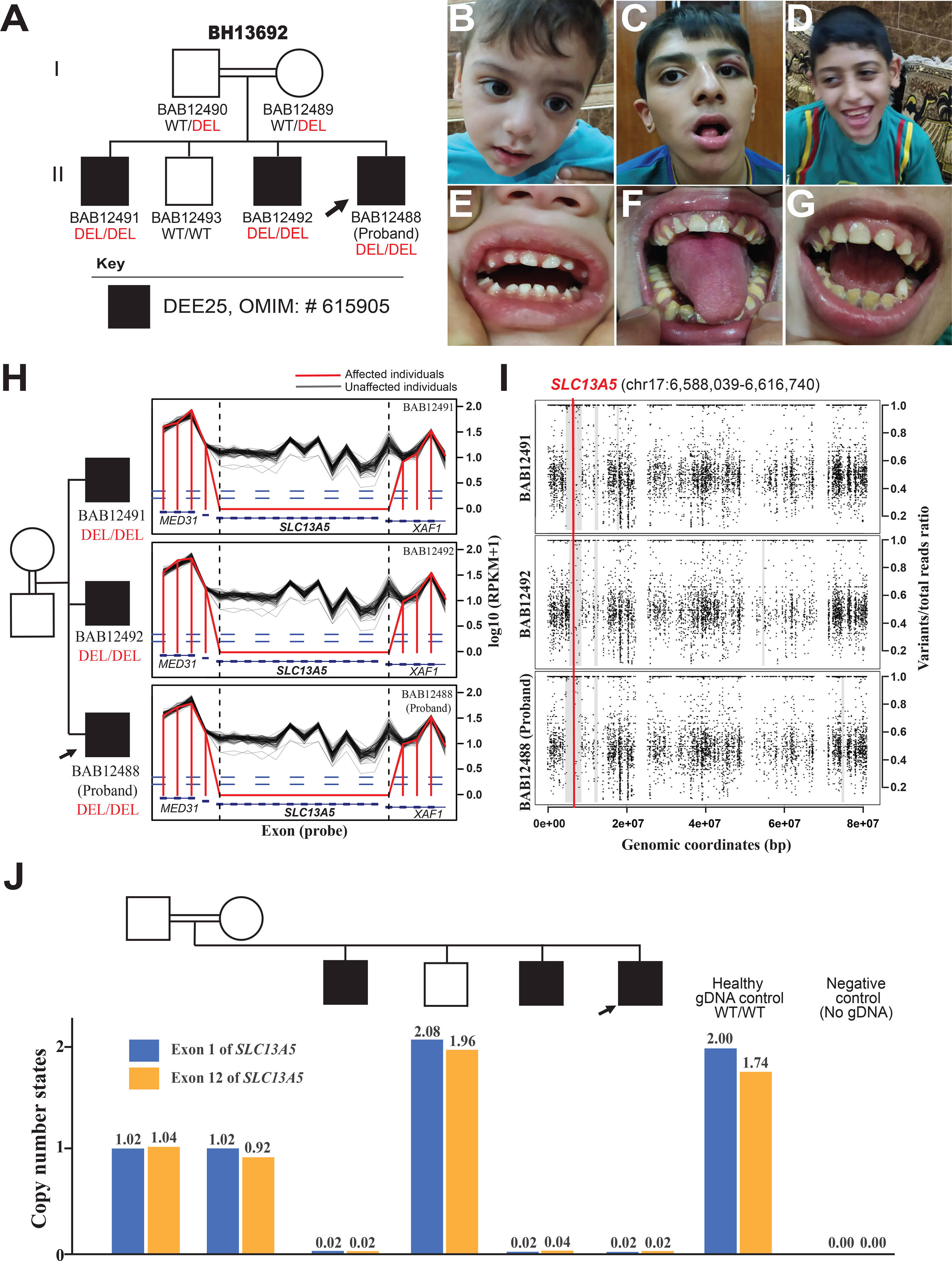
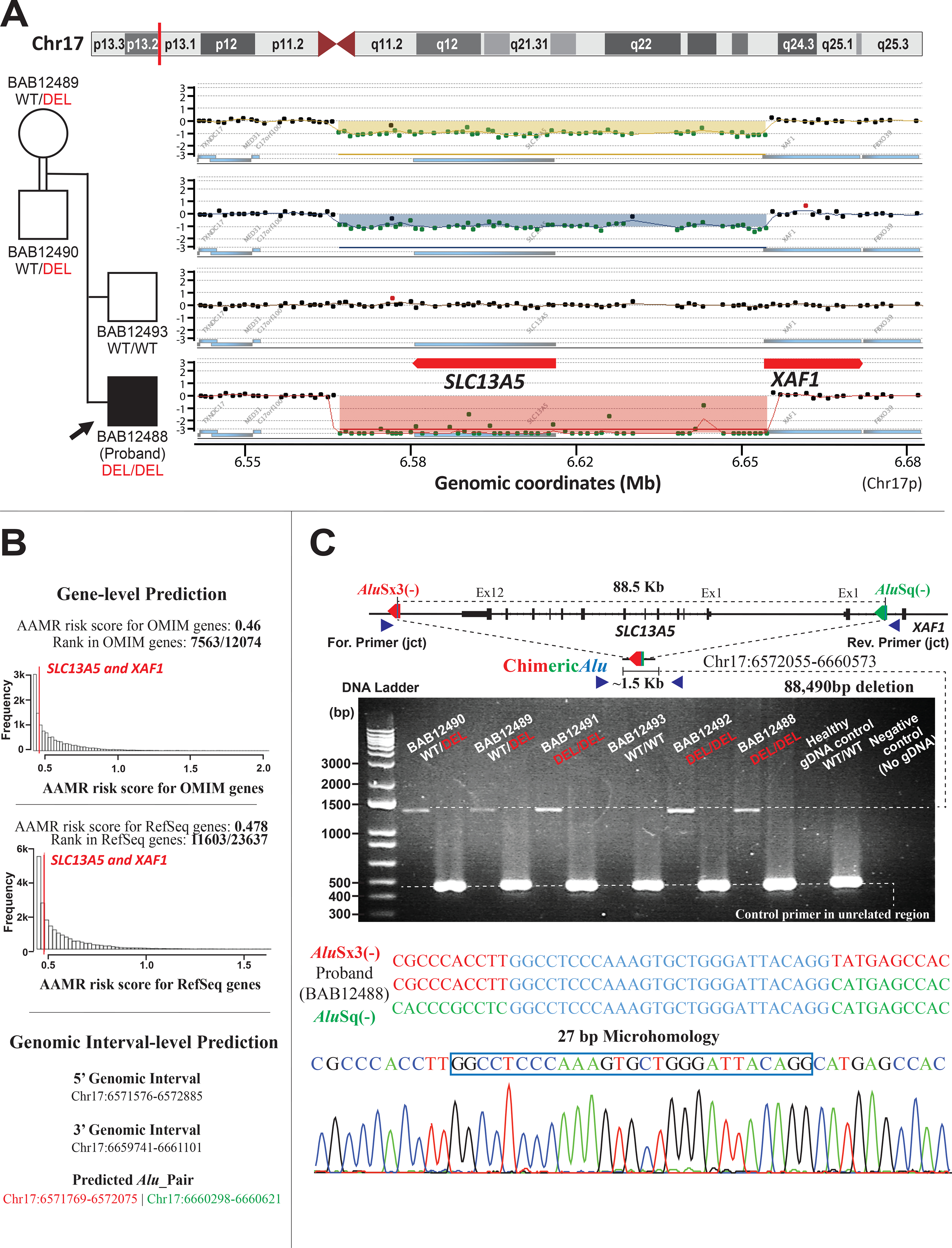
Biallelic loss-of-function (LoF) of SLC13A5 (solute carrier family 13, member 5) induced deficiency in sodium/citrate transporter (NaCT) causes autosomal recessive developmental epileptic encephalopathy 25 with hypoplastic amelogenesis imperfecta (DEE25; MIM #615905). Many pathogenic SLC13A5 single nucleotide variants (SNVs) and small indels have been described; however, no cases with copy number variants (CNVs) have been sufficiently investigated. We describe a consanguineous Iraqi family harboring an 88.5 kb homozygous deletion including SLC13A5 in Chr17p13.1. The three affected male siblings exhibit neonatal-onset epilepsy with fever-sensitivity, recurrent status epilepticus, global developmental delay/intellectual disability (GDD/ID), and other variable neurological findings as shared phenotypical features of DEE25. Two of the three affected subjects exhibit hypoplastic amelogenesis imperfecta (AI), while the proband shows no evidence of dental abnormalities or AI at 2 years of age with apparently unaffected primary dentition. Characterization of the genomic architecture at this locus revealed evidence for genomic instability generated by an Alu/Alu-mediated rearrangement; confirmed by break-point junction Sanger sequencing. This multiplex family from a distinct population elucidates the phenotypic consequence of complete LoF of SLC13A5 and illustrates the importance of read-depth-based CNV detection in comprehensive exome sequencing analysis to solve cases that otherwise remain molecularly unsolved.
Keywords: Alu/Alu-mediated rearrangement; CNV alleles; SLC13A5; developmental and epileptic encephalopathy; genomic instability; homozygous deletion.
© 2021 Wiley Periodicals LLC.
Conflict of interest statement
Conflict of interest
J.R.L. has stock ownership in 23andMe, is a paid consultant for Regeneron Pharmaceuticals, and is a co-inventor on multiple United States and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, and bacterial genomic fingerprinting. The Department of Molecular and Human Genetics at Baylor College of Medicine receives revenue from clinical genetic testing conducted at Baylor Genetics (BG) Laboratories; JRL participates in the BG scientific advisory board. Other authors have no potential conflicts to report.
Figures
References
-
- Boone PM, Yuan B, Campbell IM, Scull JC, Withers MA, Baggett BC, Beck CR, Shaw CJ, Stankiewicz P, Moretti P, Goodwin WE, Hein N, Fink JK, Seong MW, Seo SH, Park SS, Karbassi ID, Batish SD, Ordóñez-Ugalde A, … Lupski JR (2014). The Alu-rich genomic architecture of SPAST predisposes to diverse and functionally distinct disease-associated CNV alleles. American Journal of Human Genetics, 95(2), 143–161. 10.1016/j.ajhg.2014.06.014 - DOI - PMC - PubMed
-
- Carvalho CMB, Vasanth S, Shinawi M, Russell C, Ramocki MB, Brown CW, Graakjaer J, Skytte AB, Vianna-Morgante AM, Krepischi ACV, Patel GS, Immken LD, Aleck K, Lim C, Cheung SW, Rosenberg C, Katsanis N & Lupski JR (2014). Dosage changes of a segment at 17p13.1 lead to intellectual disability and microcephaly as a result of complex genetic interaction of multiple genes. American Journal of Human Genetics, 95(5), 565–578. 10.1016/j.ajhg.2014.10.006 - DOI - PMC - PubMed
-
- Dharmadhikari AV, Ghosh R, Yuan B, Liu P, Dai H, Al Masri S, Scull J, Posey JE, Jiang AH, He W, Vetrini F, Braxton AA, Ward P, Chiang T, Qu C, Gu S, Shaw CA, Smith JL, Lalani S, … Bi W (2019). Copy number variant and runs of homozygosity detection by microarrays enabled more precise molecular diagnoses in 11,020 clinical exome cases. Genome Medicine, 11(1), 1–17. 10.1186/s13073-019-0639-5 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
