

Central precocious puberty: Recent advances in understanding the aetiology and in the clinical approach
- PMID: 33797780
- PMCID: PMC8586890
- DOI: 10.1111/cen.14475
Central precocious puberty: Recent advances in understanding the aetiology and in the clinical approach
Abstract
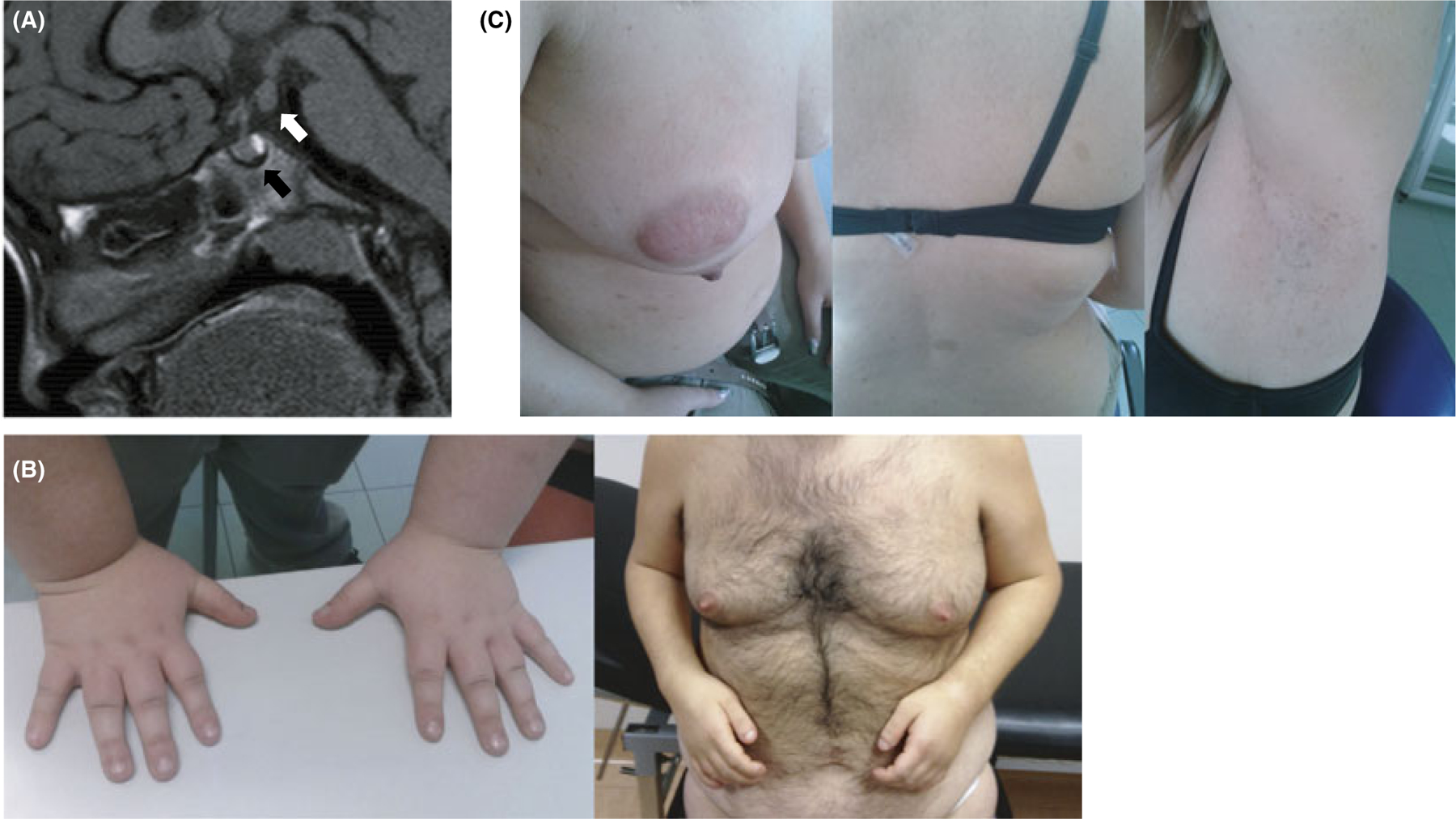
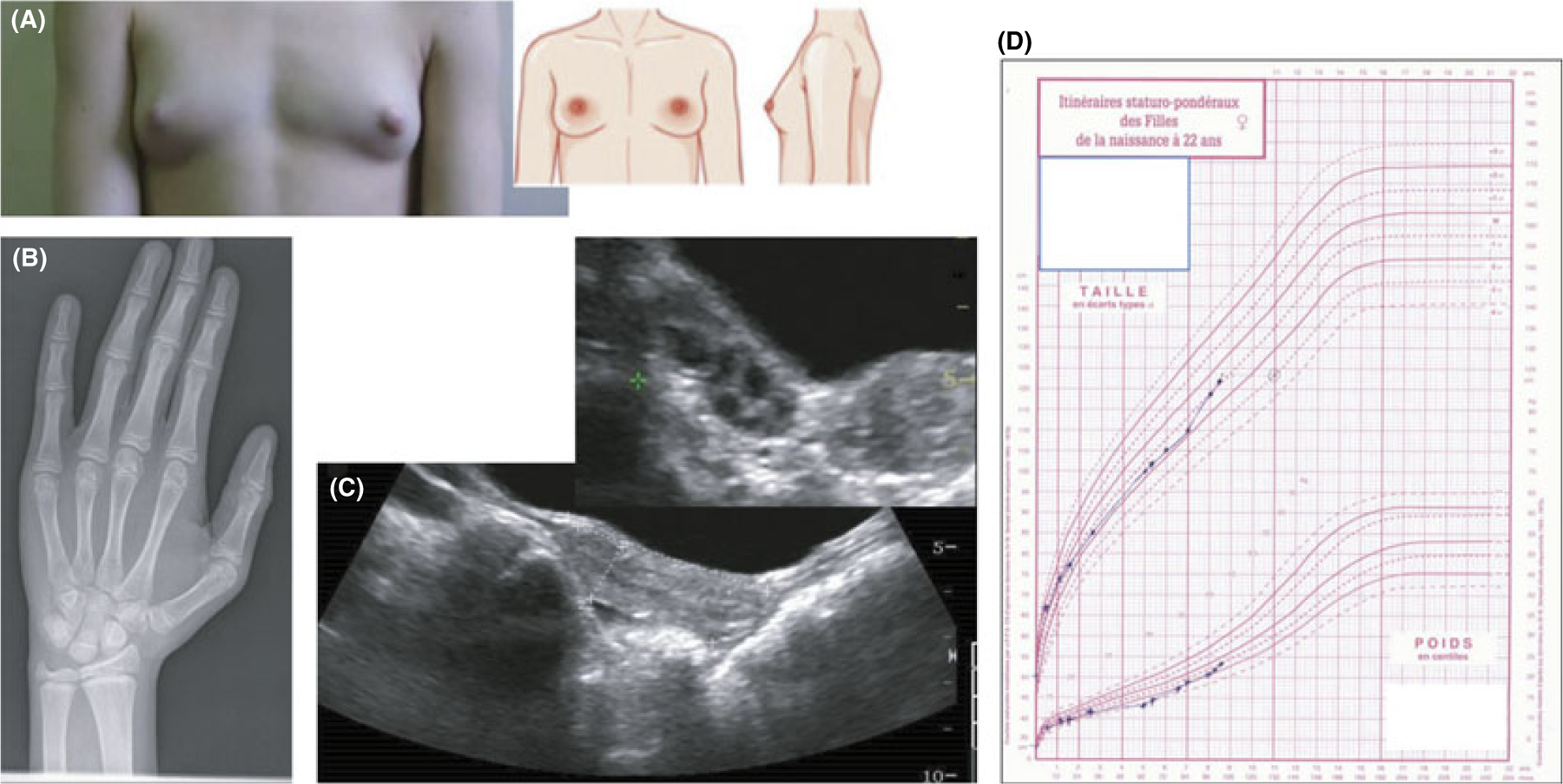
Central precocious puberty (CPP) results from early activation of the hypothalamic-pituitary-gonadal (HPG) axis. The current state of knowledge of the complex neural network acting at the level of the hypothalamus and the GnRH neuron to control puberty onset has expanded, particularly in the context of molecular interactions. Along with these advances, the knowledge of pubertal physiology and pathophysiology has also increased. This review focuses on regulatory abnormalities occurring at the hypothalamic level of the HPG axis to cause CPP. The clinical approach to diagnosis of puberty and pubertal disorders is also reviewed, with a particular focus on aetiologies of CPP. The recent identification of mutations in MKRN3 and DLK1 in familial as well sporadic forms of CPP has changed the state of the art of the approach to patients with CPP. Genetic advances have also had important repercussions beyond consideration of puberty alone. Syndromic disorders and central nervous system lesions associated with CPP are also discussed. If untreated, these conditions may lead to adverse physical, psychosocial and medical outcomes.
© 2021 John Wiley & Sons Ltd.
Conflict of interest statement
CONFLICT OF INTEREST
All the Authors declare no conflict of interest.
Figures



References
-
- Avendano MS, Vazquez MJ, Tena-Sempere M. Disentangling puberty: novel neuroendocrine pathways and mechanisms for the control of mammalian puberty. Hum Reprod Update. 2017;23(6):737–763. - PubMed
-
- Cheng CK, Leung PC. Molecular biology of gonadotropin-releasing hormone (GnRH)-I, GnRH-II, and their receptors in humans. Endocr Rev. 2005;26(2):283–306. - PubMed
-
- Seminara SB, Messager S, Chatzidaki EE, et al. The GPR54 gene as a regulator of puberty. N Engl J Med. 2003;349(17):1614–1627. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
