

Progressive myoclonus epilepsies-Residual unsolved cases have marked genetic heterogeneity including dolichol-dependent protein glycosylation pathway genes
- PMID: 33798445
- PMCID: PMC8059372
- DOI: 10.1016/j.ajhg.2021.03.013
Progressive myoclonus epilepsies-Residual unsolved cases have marked genetic heterogeneity including dolichol-dependent protein glycosylation pathway genes
Abstract
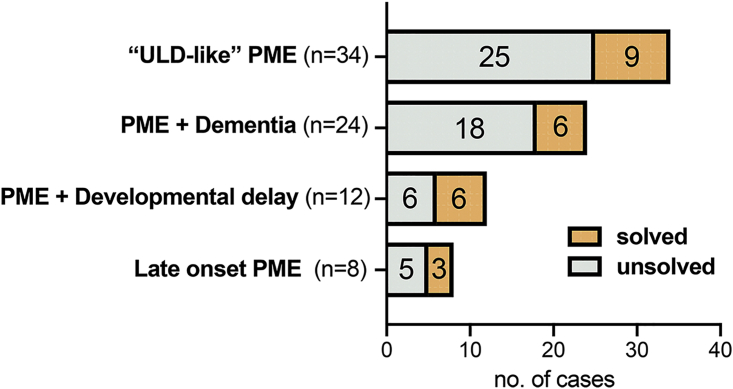
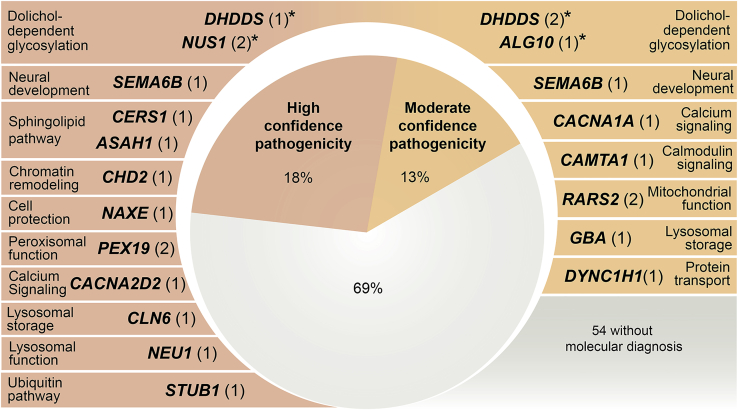
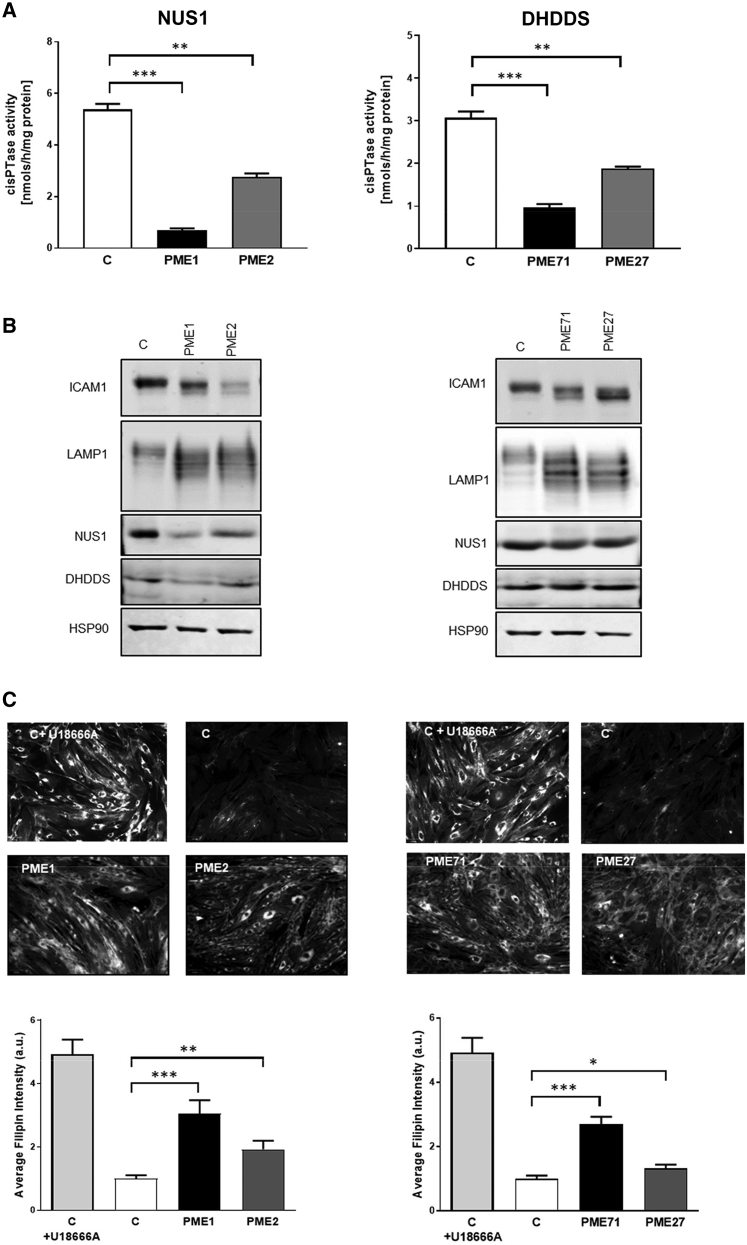
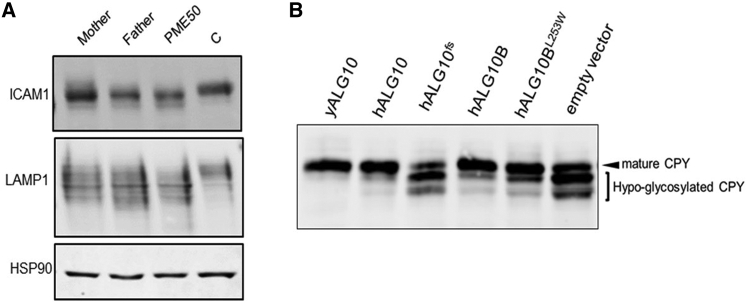
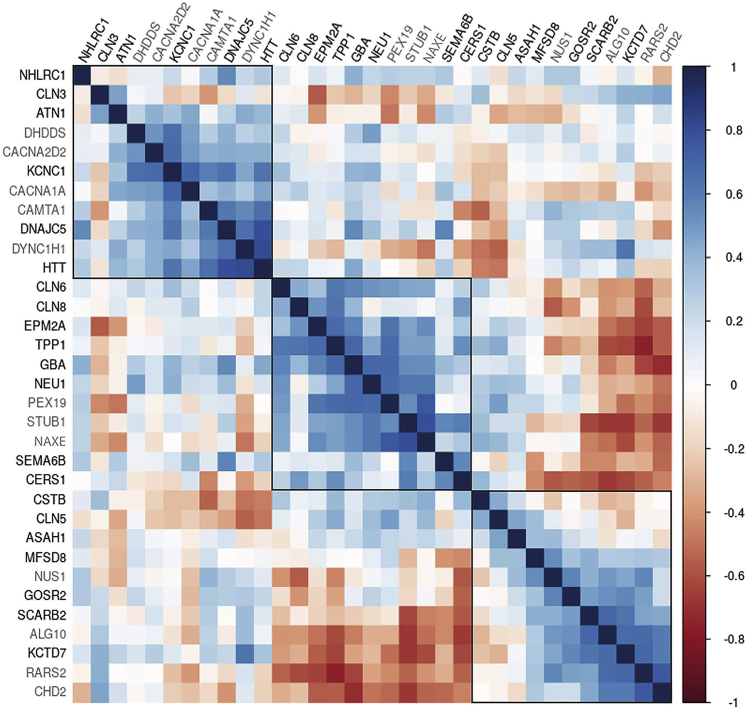
Progressive myoclonus epilepsies (PMEs) comprise a group of clinically and genetically heterogeneous rare diseases. Over 70% of PME cases can now be molecularly solved. Known PME genes encode a variety of proteins, many involved in lysosomal and endosomal function. We performed whole-exome sequencing (WES) in 84 (78 unrelated) unsolved PME-affected individuals, with or without additional family members, to discover novel causes. We identified likely disease-causing variants in 24 out of 78 (31%) unrelated individuals, despite previous genetic analyses. The diagnostic yield was significantly higher for individuals studied as trios or families (14/28) versus singletons (10/50) (OR = 3.9, p value = 0.01, Fisher's exact test). The 24 likely solved cases of PME involved 18 genes. First, we found and functionally validated five heterozygous variants in NUS1 and DHDDS and a homozygous variant in ALG10, with no previous disease associations. All three genes are involved in dolichol-dependent protein glycosylation, a pathway not previously implicated in PME. Second, we independently validate SEMA6B as a dominant PME gene in two unrelated individuals. Third, in five families, we identified variants in established PME genes; three with intronic or copy-number changes (CLN6, GBA, NEU1) and two very rare causes (ASAH1, CERS1). Fourth, we found a group of genes usually associated with developmental and epileptic encephalopathies, but here, remarkably, presenting as PME, with or without prior developmental delay. Our systematic analysis of these cases suggests that the small residuum of unsolved cases will most likely be a collection of very rare, genetically heterogeneous etiologies.
Keywords: dolichol-dependent glycosylation; epilepsy genetics; progressive myoclonus epilepsy; whole-exome sequencing.
Copyright © 2021 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
M.M. is employed by Blueprint Genetics. All other authors declare no competing interests.
Figures
References
-
- Berkovic S.F., Andermann F., Carpenter S., Wolfe L.S. Progressive myoclonus epilepsies: specific causes and diagnosis. N. Engl. J. Med. 1986;315:296–305. - PubMed
-
- Ramachandran N., Girard J.M., Turnbull J., Minassian B.A. The autosomal recessively inherited progressive myoclonus epilepsies and their genes. Epilepsia. 2009;50(Suppl 5):29–36. - PubMed
-
- Kollmann K., Uusi-Rauva K., Scifo E., Tyynelä J., Jalanko A., Braulke T. Cell biology and function of neuronal ceroid lipofuscinosis-related proteins. Biochim. Biophys. Acta. 2013;1832:1866–1881. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
