

Neuropathology of Animal Prion Diseases
- PMID: 33801117
- PMCID: PMC8004141
- DOI: 10.3390/biom11030466
Neuropathology of Animal Prion Diseases
Abstract
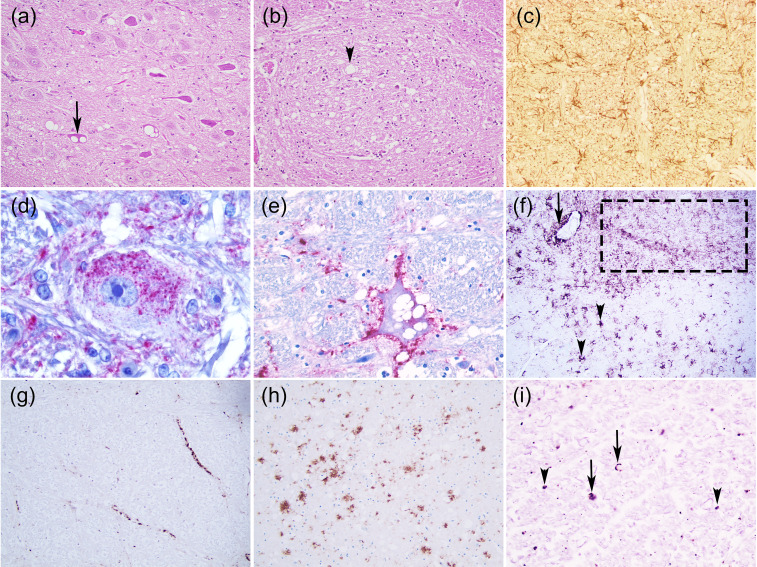
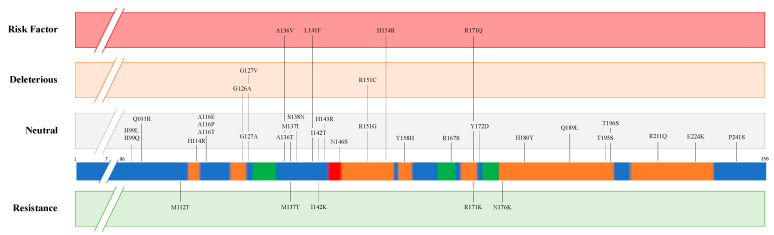
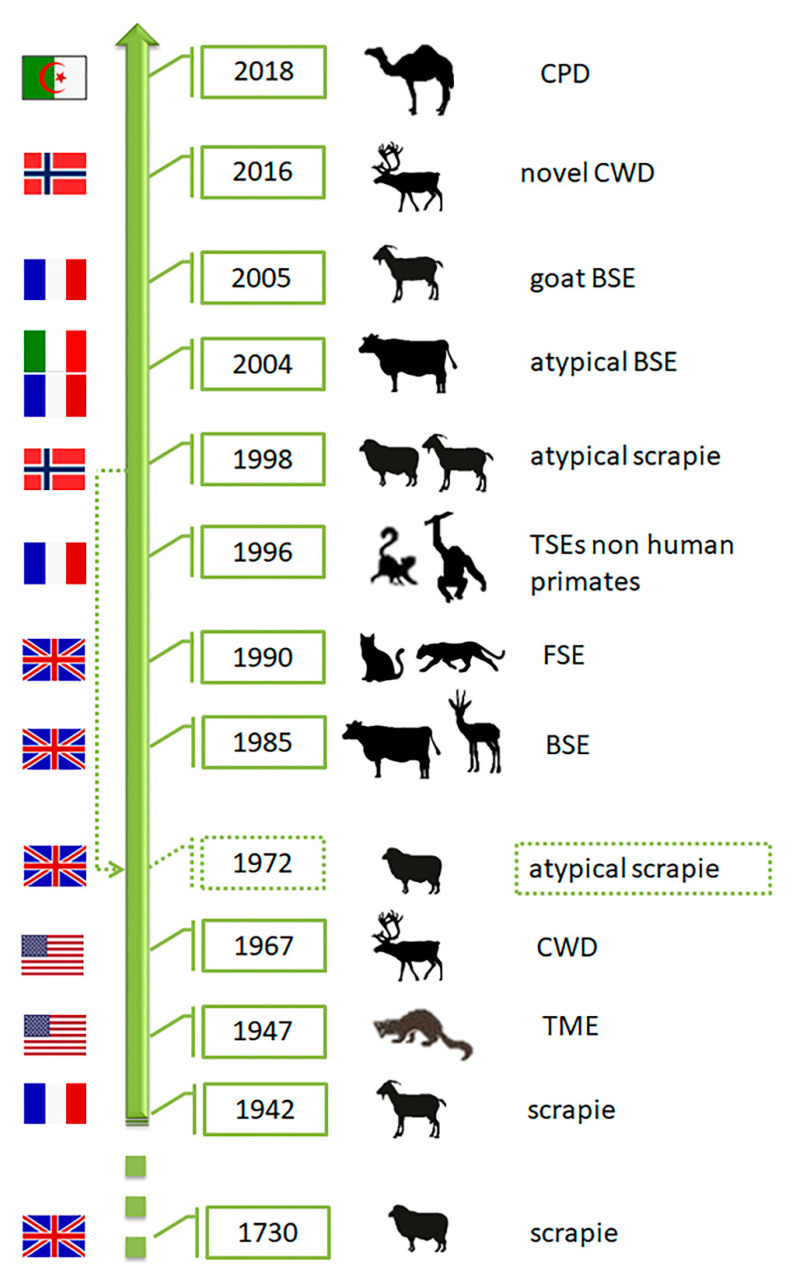
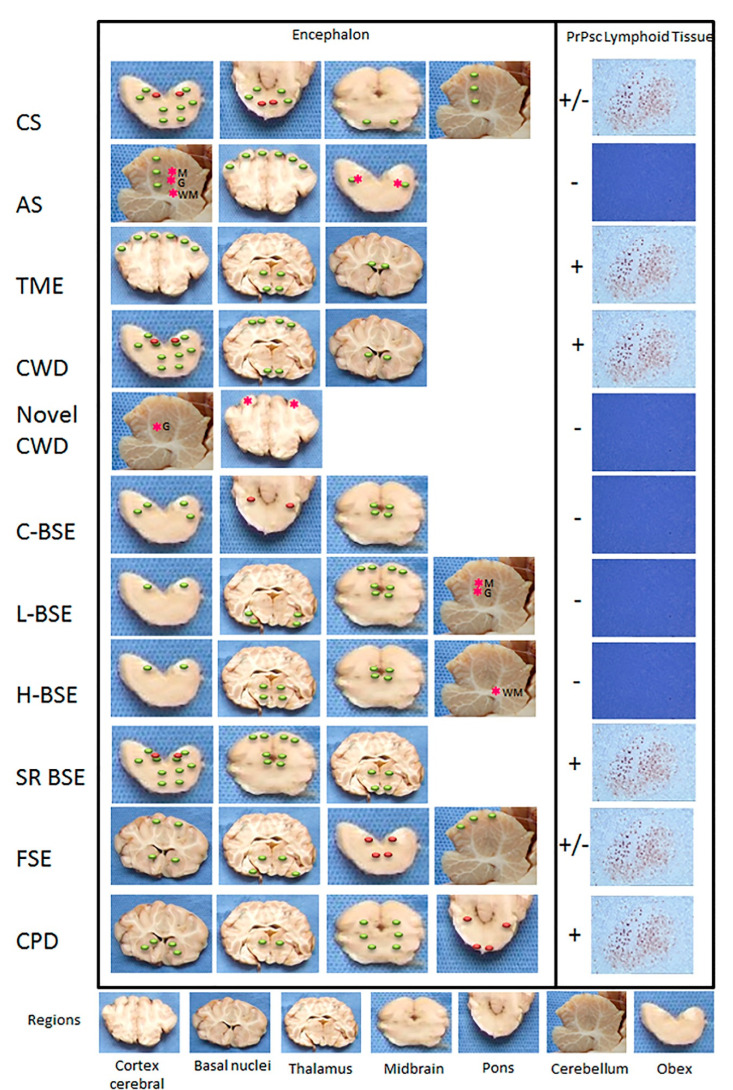
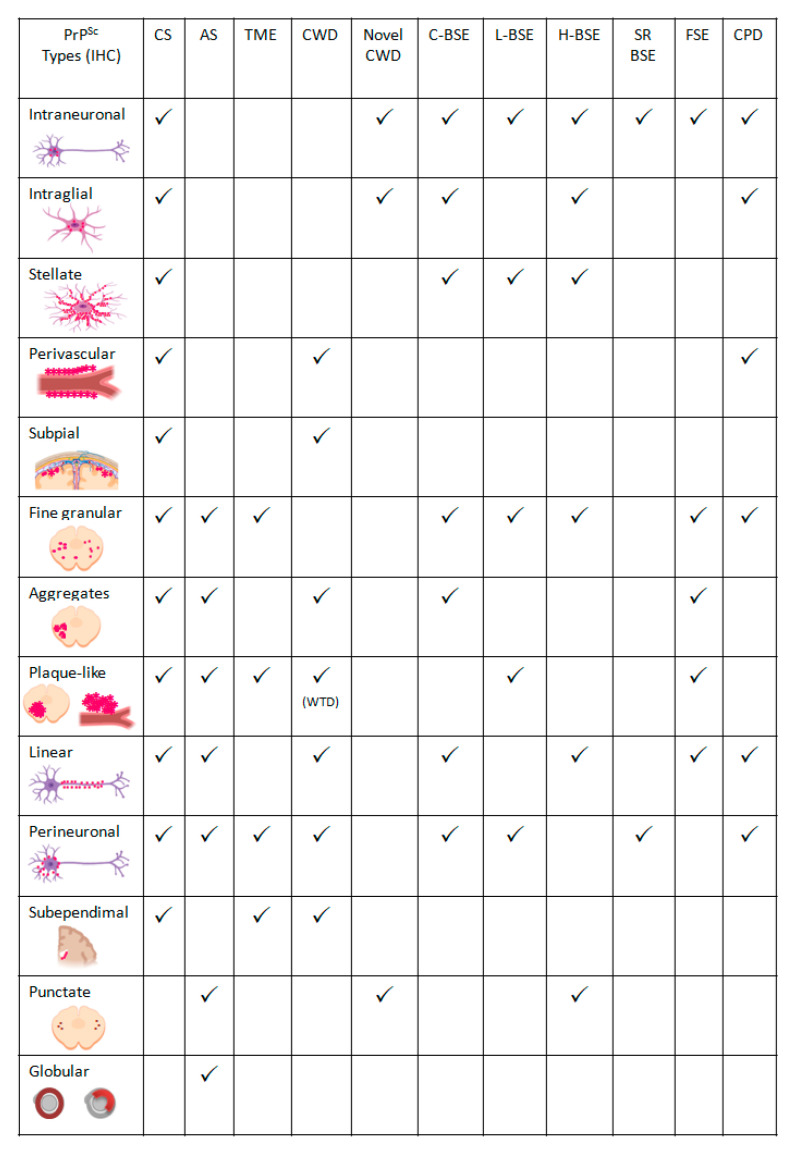
Transmissible Spongiform Encephalopathies (TSEs) or prion diseases are a fatal group of infectious, inherited and spontaneous neurodegenerative diseases affecting human and animals. They are caused by the conversion of cellular prion protein (PrPC) into a misfolded pathological isoform (PrPSc or prion- proteinaceous infectious particle) that self-propagates by conformational conversion of PrPC. Yet by an unknown mechanism, PrPC can fold into different PrPSc conformers that may result in different prion strains that display specific disease phenotype (incubation time, clinical signs and lesion profile). Although the pathways for neurodegeneration as well as the involvement of brain inflammation in these diseases are not well understood, the spongiform changes, neuronal loss, gliosis and accumulation of PrPSc are the characteristic neuropathological lesions. Scrapie affecting small ruminants was the first identified TSE and has been considered the archetype of prion diseases, though atypical and new animal prion diseases continue to emerge highlighting the importance to investigate the lesion profile in naturally affected animals. In this report, we review the neuropathology and the neuroinflammation of animal prion diseases in natural hosts from scrapie, going through the zoonotic bovine spongiform encephalopathy (BSE), the chronic wasting disease (CWD) to the newly identified camel prion disease (CPD).
Keywords: animal TSE; gene PRNP; neuroinflammation; neuropathology; prion; spongiform degeneration.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

References
-
- VanDeVelde M., Higgins R.J., Oevermann A. Veterinary Neuropathology: Essentials of Theory and Practice. Wiley-Blackwell; Oxford, UK: 2012.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
