

Expanding the β-III Spectrin-Associated Phenotypes toward Non-Progressive Congenital Ataxias with Neurodegeneration
- PMID: 33801522
- PMCID: PMC7958857
- DOI: 10.3390/ijms22052505
Expanding the β-III Spectrin-Associated Phenotypes toward Non-Progressive Congenital Ataxias with Neurodegeneration
Abstract
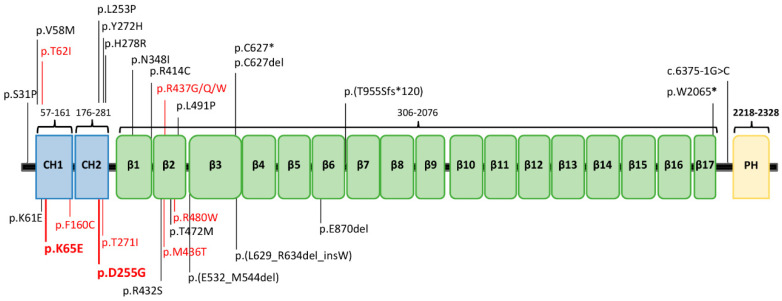
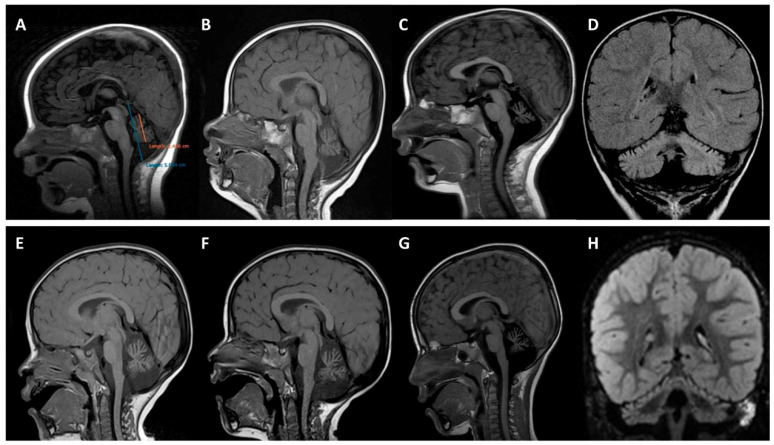
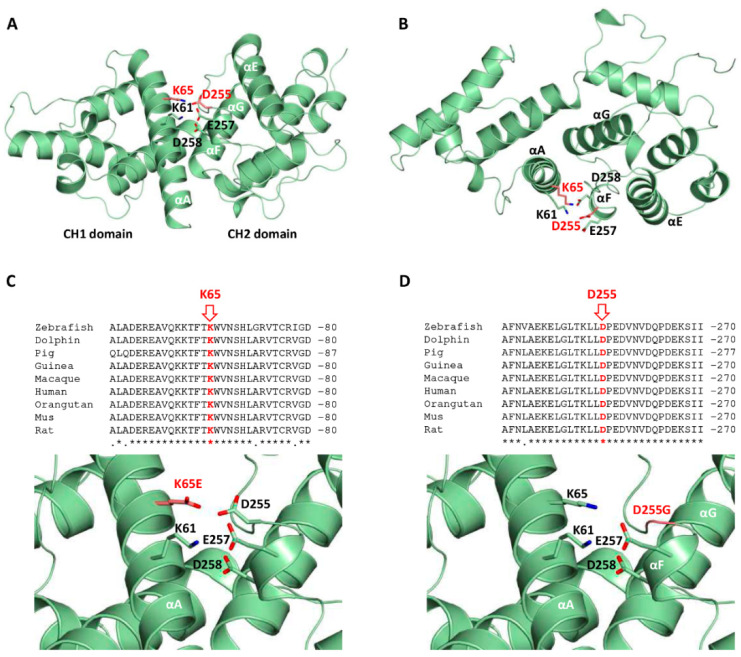
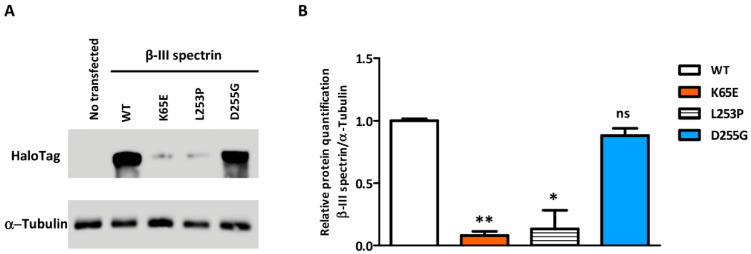
(1) Background: A non-progressive congenital ataxia (NPCA) phenotype caused by β-III spectrin (SPTBN2) mutations has emerged, mimicking spinocerebellar ataxia, autosomal recessive type 14 (SCAR14). The pattern of inheritance, however, resembles that of autosomal dominant classical spinocerebellar ataxia type 5 (SCA5). (2) Methods: In-depth phenotyping of two boys studied by a customized gene panel. Candidate variants were sought by structural modeling and protein expression. An extensive review of the literature was conducted in order to better characterize the SPTBN2-associated NPCA. (3) Results: Patients exhibited an NPCA with hypotonia, developmental delay, cerebellar syndrome, and cognitive deficits. Both probands presented with progressive global cerebellar volume loss in consecutive cerebral magnetic resonance imaging studies, characterized by decreasing midsagittal vermis relative diameter measurements. Cortical hyperintensities were observed on fluid-attenuated inversion recovery (FLAIR) images, suggesting a neurodegenerative process. Each patient carried a novel de novo SPTBN2 substitution: c.193A > G (p.K65E) or c.764A > G (p.D255G). Modeling and protein expression revealed that both mutations might be deleterious. (4) Conclusions: The reported findings contribute to a better understanding of the SPTBN2-associated phenotype. The mutations may preclude proper structural organization of the actin spectrin-based membrane skeleton, which, in turn, is responsible for the underlying disease mechanism.
Keywords: SPTBN2 gene; neurodegeneration; non-progressive congenital ataxia; β-III spectrin.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Lise S., Clarkson Y., Perkins E., Kwasniewska A., Sadighi Akha E., Schnekenberg R.P., Suminaite D., Hope J., Baker I., Gregory L., et al. Recessive mutations in SPTBN2 implicate beta-III spectrin in both cognitive and motor development. PLoS Genet. 2012;8:e1003074. doi: 10.1371/journal.pgen.1003074. - DOI - PMC - PubMed
-
- Sun M., Johnson A.K., Nelakuditi V., Guidugli L., Fischer D., Arndt K., Ma L., Sandford E., Shakkottai V., Boycott K., et al. Targeted exome analysis identifies the genetic basis of disease in over 50% of patients with a wide range of ataxia-related phenotypes. Genet. Med. 2018;21:195–206. doi: 10.1038/s41436-018-0007-7. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
