

Clinical and Genetic Characteristics of Patients with Mild Hyperphenylalaninemia Identified by Newborn Screening Program in Japan
- PMID: 33803550
- PMCID: PMC8006226
- DOI: 10.3390/ijns7010017
Clinical and Genetic Characteristics of Patients with Mild Hyperphenylalaninemia Identified by Newborn Screening Program in Japan
Abstract
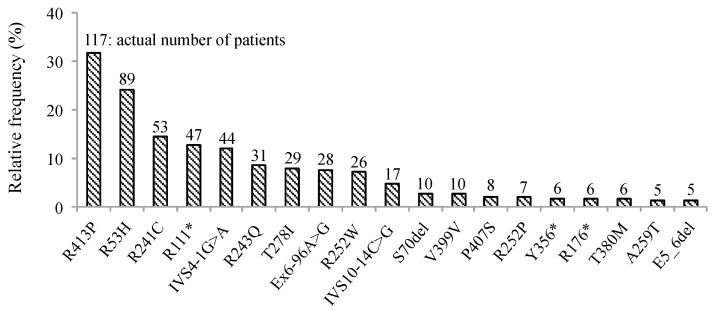
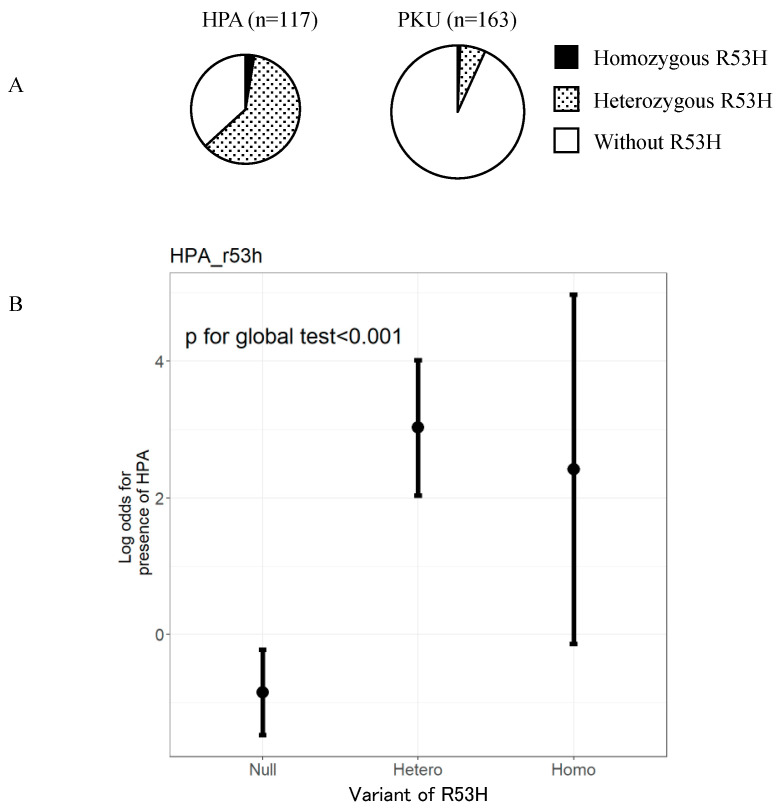
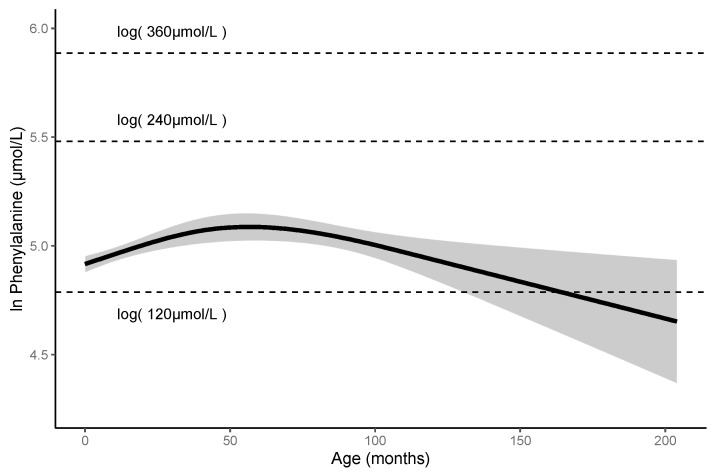
Phenylketonuria (PKU) and hyperphenylalaninemia (HPA), both identified in newborn screening, are attributable to variants in PAH. Reportedly, the p.R53H(c.158G>A) variant is common in patients with HPA in East Asia. Here, we aimed to define the association between p.R53H and HPA phenotype, and study the long-term outcome of patients with HPA carrying p.R53H. We retrospectively reviewed the genotype in 370 patients detected by newborn screening, and identified the phenotype in 280 (117, HPA; 163, PKU). p.R413P(c.1238G>C) was the most frequently found (n = 117, 31.6%) variant, followed by p.R53H (n = 89, 24.1%). The odds ratio for heterozygous p.R53H to cause HPA was 48.3 (95% CI 19.410-120.004). Furthermore, we assessed the non-linear association between the phenylalanine (Phe) value and elapsed time using the follow-up data of the blood Phe levels of 73 patients with HPA carrying p.R53H. The predicted levels peaked at 161.9 μmol (95% CI 152.088-172.343) at 50-60 months of age and did not exceed 360 μmol/L during the 210-month long observation period. The findings suggest that patients with HPA, carrying p.R53H, do not need frequent Phe monitoring as against those with PKU. Our study provides convincing evidence to determine clinical management of patients detected through newborn screening in Japan.
Keywords: genetic analysis; genotype–phenotype correlation; hyperphenylalaninemia; neonatal screening; phenylalanine hydroxylase; phenylketonuria.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Guldberg P., Rey F., Zschocke J., Romano V., François B., Michiels L., Ullrich K., Hoffmann G.F., Burgard P., Schmidt H., et al. A European Multicenter Study of Phenylalanine Hydroxylase Deficiency: Classification of 105 Mutations and a General System for Genotype-Based Prediction of Metabolic Phenotype. Am. J. Hum. Genet. 1998;63:71–79. doi: 10.1086/301920. - DOI - PMC - PubMed
-
- Van Wegberg A.M.J., Macdonald A., Ahring K., BéLanger-Quintana A., Blau N., Bosch A.M., Burlina A., Campistol J., Feillet F., Giżewska M., et al. The complete European guidelines on phenylketonuria: Diagnosis and treatment. Orphanet J. Rare Dis. 2017;12:1–56. doi: 10.1186/s13023-017-0685-2. - DOI - PMC - PubMed
-
- Camp K.M., Parisi M.A., Acosta P.B., Berry G.T., Bilder D.A., Blau N., Bodamer O.A., Brosco J.P., Brown C.S., Burlina A.B., et al. Phenylketonuria Scientific Review Conference: State of the science and future research needs. Mol. Genet. Metab. 2014;112:87–122. doi: 10.1016/j.ymgme.2014.02.013. - DOI - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
