

LRRK2 at the Crossroad of Aging and Parkinson's Disease
- PMID: 33805527
- PMCID: PMC8066012
- DOI: 10.3390/genes12040505
LRRK2 at the Crossroad of Aging and Parkinson's Disease
Abstract
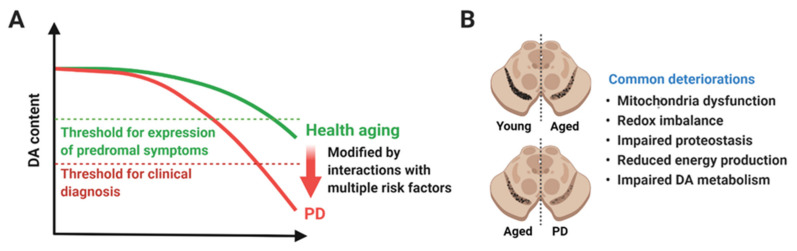
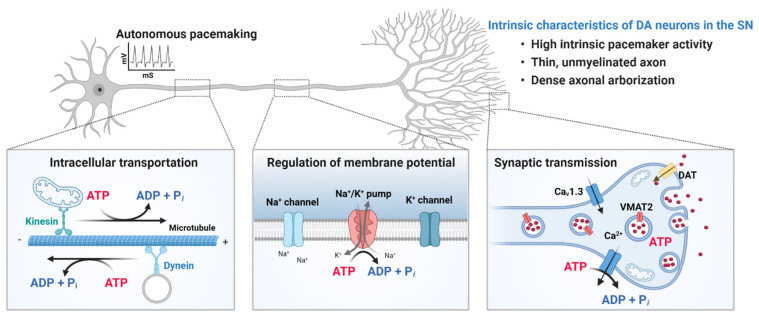
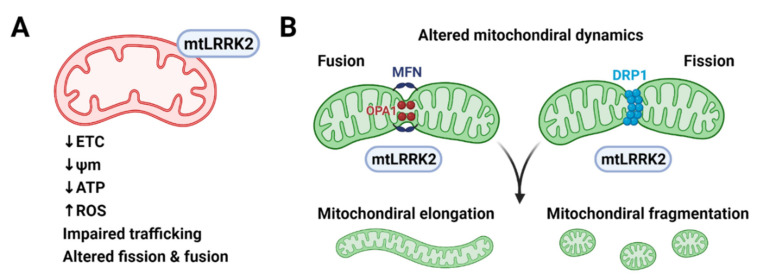
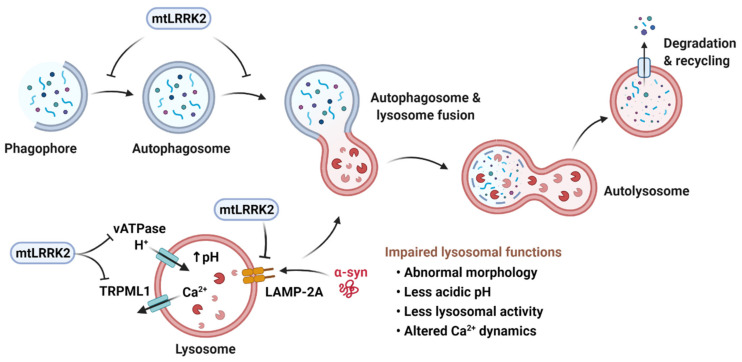
Parkinson's disease (PD) is a heterogeneous neurodegenerative disease characterized by the progressive loss of dopaminergic neurons in the substantia nigra pars compacta and the widespread occurrence of proteinaceous inclusions known as Lewy bodies and Lewy neurites. The etiology of PD is still far from clear, but aging has been considered as the highest risk factor influencing the clinical presentations and the progression of PD. Accumulating evidence suggests that aging and PD induce common changes in multiple cellular functions, including redox imbalance, mitochondria dysfunction, and impaired proteostasis. Age-dependent deteriorations in cellular dysfunction may predispose individuals to PD, and cellular damages caused by genetic and/or environmental risk factors of PD may be exaggerated by aging. Mutations in the LRRK2 gene cause late-onset, autosomal dominant PD and comprise the most common genetic causes of both familial and sporadic PD. LRRK2-linked PD patients show clinical and pathological features indistinguishable from idiopathic PD patients. Here, we review cellular dysfunctions shared by aging and PD-associated LRRK2 mutations and discuss how the interplay between the two might play a role in PD pathologies.
Keywords: LRRK2; Parkinson’s disease; ROS; aging; autophagy; lysosome; mitochondria.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
